
Abstract
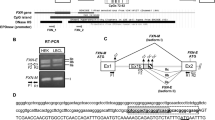
Friedreich's ataxia (FRDA), the most common autosomal recessively inherited ataxia, is due to a homozygous GAA triplet repeat expansion in the first intron of the FRDA gene in about 96% of patients. Approximately 4% of FRDA patients are compound heterozygotes with a GAA repeat expansion in one allele and a point mutation in the coding region of the second allele. To reinvestigate the mutation spectrum, we searched for mutations including exon deletions in six patients heterozygous for the GAA repeat expansion and found two unknown missense mutations, p.Asn146Lys and p.Leu186Arg, in trans to the expanded FRDA allele. Interestingly, we detected a heterozygous 2776 bp deletion including exon 5a in one of our patients. This deletion removes 50 of the 210 residues of the frataxin. Furthermore, since no FRDA case with two-point mutations is known, we screened eight patients with FRDA phenotype but GAA alleles within the normal range but did not reveal a mutation within the FRDA gene. In addition, DNA polymorphisms have been found in four out of 100 control individuals in this study.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Campuzano V, Montermini L, Molto MD et al: Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996; 271: 1423–1427.
Montermini L, Andermann E, Labuda M et al: The Friedreich ataxia GAA triplet repeat: premutation and normal alleles. Hum Mol Genet 1997; 6: 1261–1266.
Cossee M, Schmitt M, Campuzano V et al: Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci USA 1997; 94: 7452–7457.
Zühlke CH, Laccone F, Cossee M, Kohlschütter A, Koenig M, Schwinger E : Mutation of the start codon in the FRDA1 gene: linkage analysis of three pedigrees with the ATG to ATT transversion points to a unique common ancestor. Hum Genet 1998; 103: 102–105.
Hedrich K, Kann M, Lanthaler AJ et al: The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset Parkinsonism. Hum Mol Genet 2001; 10: 1649–1656.
Koutnikova H, Campuzano V, Foury F, Dolle P, Cazzalini O, Koenig M : Studies of human, mouse and yeast homologues indicate a mitochondrial function of frataxin. Nat Genet 1997; 16: 345–351.
Pandolfo M, Montermini L : Prenatal diagnosis of Friedreich ataxia. Prenatal Diagn 1998; 18: 831–833.
Acknowledgements
We thank U Gehlken and J Atici for excellent technical help. We thank all patients for providing blood samples for scientific research as well as the German Heredo-Ataxia Society (DHAG), whose cooperation and support is essential in our work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zühlke, C., Dalski, A., Habeck, M. et al. Extension of the mutation spectrum in Friedreich's ataxia: detection of an exon deletion and novel missense mutations. Eur J Hum Genet 12, 979–982 (2004). https://doi.org/10.1038/sj.ejhg.5201257
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.ejhg.5201257
Keywords
This article is cited by
-
DNA methylation in Friedreich ataxia silences expression of frataxin isoform E
Scientific Reports (2022)
-
Friedreich ataxia is not only a GAA repeats expansion disorder: implications for molecular testing and counselling
Journal of Applied Genetics (2016)
-
Friedreich ataxia in Norway – an epidemiological, molecular and clinical study
Orphanet Journal of Rare Diseases (2015)
-
Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells
Pflügers Archiv - European Journal of Physiology (2014)
-
Klinik und Genetik der rezessiven Ataxien
Der Nervenarzt (2011)
