
Abstract
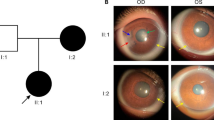
Anterior segment dysgenesis (ASD) encompasses a broad spectrum of developmental conditions affecting anterior ocular structures and associated with an increased risk for glaucoma. Various systemic anomalies are often observed in ASD conditions such as Axenfeld-Rieger syndrome (ARS) and De Hauwere syndrome. We report DNA sequencing and copy number analysis of PITX2 and FOXC1 in 76 patients with syndromic or isolated ASD and related conditions. PITX2 mutations and deletions were found in 24 patients with dental and/or umbilical anomalies seen in all. Seven PITX2-mutant alleles were novel including c.708_730del, the most C-terminal mutation reported to date. A second case of deletion of the distant upstream but not coding region of PITX2 was identified, highlighting the importance of this recently discovered mechanism for ARS. FOXC1 deletions were observed in four cases, three of which demonstrated hearing and/or heart defects, including a patient with De Hauwere syndrome; no nucleotide mutations in FOXC1 were identified. Review of the literature identified several other patients with 6p25 deletions and features of De Hauwere syndrome. The 1.3-Mb deletion of 6p25 presented here defines the critical region for this phenotype and includes the FOXC1, FOXF2, and FOXQ1 genes. In summary, PITX2 or FOXC1 disruptions explained 63% of ARS and 6% of other ASD in our cohort; all affected patients demonstrated additional systemic defects with PITX2 mutations showing a strong association with dental and/or umbilical anomalies and FOXC1 with heart and hearing defects. FOXC1 deletion was also found to be associated with De Hauwere syndrome.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Sowden JC : Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond) 2007; 21: 1310–1318.
Reis LM, Semina EV : Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol 2011; 22: 314–324.
Rieger H : Verlagerung und schlitzform der pupille mit hypoplasie des irisvorderblattes. Z Augenheilkd 1934; 84: 98–103.
Alward WL : Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol 2000; 130: 107–115.
Tümer Z, Bach-Holm D : Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet 2009; 17: 1527–1539.
Semina EV, Reiter R, Leysens NJ et al: Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996; 14: 392–399.
Mears AJ, Jordan T, Mirzayans F et al: Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet 1998; 63: 1316–1328.
Nishimura DY, Swiderski RE, Alward WL et al: The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet 1998; 19: 140–147.
Hjalt TA, Semina EV : Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med 2005; 7: 1–17.
D'haene B, Meire F, Claerhout I et al: Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Invest Ophthalmol Vis Sci 2011; 52: 324–333.
Strungaru MH, Dinu I, Walter MA : Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci 2007; 48: 228–237.
Kozlowski K, Walter MA : Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum Mol Genet 2000; 9: 2131–2139.
Espinoza HM, Cox CJ, Semina EV, Amendt BA : A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum Mol Genet 2002; 11: 743–753.
Maciolek NL, Alward WL, Murray JC, Semina EV, McNally MT : Analysis of RNA splicing defects in PITX2 mutants supports a gene dosage model of Axenfeld-Rieger syndrome. BMC Med Genet 2006; 7: 59.
Priston M, Kozlowski K, Gill D et al: Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum Mol Genet 2001; 10: 1631–1638.
Saadi I, Semina EV, Amendt BA et al: Identification of a dominant negative homeodomain mutation in Rieger syndrome. J Biol Chem 2001; 276: 23034–23041.
Saadi I, Toro R, Kuburas A, Semina E, Murray JC, Russo AF : An unusual class of PITX2 mutations in Axenfeld-Rieger syndrome. Birth Defects Res A Clin Mol Teratol 2006; 76: 175–181.
Lines MA, Kozlowski K, Kulak SC et al: Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci 2004; 45: 828–833.
Volkmann BA, Zinkevich NS, Mustonen A et al: Potential novel mechanism for axenfeld-rieger syndrome: deletion of a distant region containing regulatory elements of PITX2. Invest Ophthalmol Vis Sci 2011; 52: 1450–1459.
Lehmann OJ, Ebenezer ND, Ekong R et al: Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Invest Ophthalmol Vis Sci 2002; 43: 1843–1849.
De Hauwere RC, Leroy JG, Adriaenssens K, Van Heule R : Iris dysplasia, orbital hypertelorism, and psychomotor retardation: a dominantly inherited developmental syndrome. J Pediatr 1973; 82: 679–681.
Lowry RB, Gould DB, Walter MA, Savage PR : Absence of PITX2, BARX1, and FOXC1 mutations in De Hauwere syndrome (Axenfeld-Rieger anomaly, hydrocephaly, hearing loss): a 25-year follow up. Am J Med Genet A 2007; 143A: 1227–1230.
Bremond-Gignac D, Bitoun P, Reis LM, Copin H, Murray JC, Semina EV : Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol Vis 2010; 16: 1705–1711.
Kaur K, Ragge NK, Ragoussis J : Molecular analysis of FOXC1 in subjects presenting with severe developmental eye anomalies. Mol.Vis 2009; 15: 1366–1373.
Exome Variant Server: NHLBI Exome Sequencing Project (ESP). Seattle, WA, November 2011.
Reis LM, Tyler RC, Schilter KF et al: BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum Genet 2011; 130: 495–504.
Divina P, Kvitkovicova A, Buratti E, Vorechovsky I : Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur J Hum Genet 2009; 17: 759–765.
Nagy E, Maquat LE : A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998; 23: 198–199.
Karadeniz NN, Kocak-Midillioglu I, Erdogan D, Bokesoy I : Is SHORT syndrome another phenotypic variation of PITX2? Am J Med Genet A 2004; 130A: 406–409.
Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T : 3' deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci USA 2000; 97: 13755–13759.
Borges AS, Susanna R, Carani JC et al: Genetic analysis of PITX2 and FOXC1 in Rieger Syndrome patients from Brazil. J Glaucoma 2002; 11: 51–56.
Gould DB, Jaafar MS, Addison MK et al: Phenotypic and molecular assessment of seven patients with 6p25 deletion syndrome: relevance to ocular dysgenesis and hearing impairment. BMC Med Genet 2004; 5: 17.
DeScipio C : The 6p subtelomere deletion syndrome. Am J Med Genet C Semin Med Genet 2007; 145C: 377–382.
Mirza G, Williams RR, Mohammed S et al: Refined genotype-phenotype correlations in cases of chromosome 6p deletion syndromes. Eur J Hum Genet 2004; 12: 718–728.
Maclean K, Smith J, Heaps L et al: Axenfeld-Rieger malformation and distinctive facial features: clues to a recognizable 6p25 microdeletion syndrome. Am J Med Genet A 2005; 132: 381–385.
Kannu P, Oei P, Slater HR, Khammy O, Aftimos S : Epiphyseal dysplasia and other skeletal anomalies in a patient with the 6p25 microdeletion syndrome. Am J Med Genet A 2006; 140: 1955–1959.
Martinez-Glez V, Lorda-Sanchez I, Ramirez JM et al: Clinical presentation of a variant of Axenfeld-Rieger syndrome associated with subtelomeric 6p deletion. Eur J Med Genet 2007; 50: 120–127.
Bedoyan JK, Lesperance MM, Ackley T, Iyer RK, Innis JW, Misra VK : A complex 6p25 rearrangement in a child with multiple epiphyseal dysplasia. Am J Med Genet A 2011; 155A: 154–163.
Anderlid BM, Schoumans J, Hallqvist A et al: Cryptic subtelomeric 6p deletion in a girl with congenital malformations and severe language impairment. Eur J Hum Genet 2003; 11: 89–92.
Hong HK, Lass JH, Chakravarti A : Pleiotropic skeletal and ocular phenotypes of the mouse mutation congenital hydrocephalus (ch/Mf1) arise from a winged helix/forkhead transcriptionfactor gene. Hum Mol Genet 1999; 8: 625–637.
Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL : The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 1998; 93: 985–996.
Wang T, Tamakoshi T, Uezato T et al: Forkhead transcription factor Foxf2 (LUN)-deficient mice exhibit abnormal development of secondary palate. Dev Biol 2003; 259: 83–94.
McKeone R, Vieira H, Gregory-Evans K, Gregory-Evans CY, Denny P : Foxf2: a novel locus for anterior segment dysgenesis adjacent to the foxc1 gene. PLoS One 2011; 6: e25489.
Hong HK, Noveroske JK, Headon DJ et al: The winged helix/forkhead transcription factor Foxq1 regulates differentiation of hair in satin mice. Genesis 2001; 29: 163–171.
Planchart A, Mattingly CJ : 2,3,7,8-Tetrachlorodibenzo-p-dioxin upregulates FoxQ1b in zebrafish jaw primordium. Chem Res Toxicol 2010; 23: 480–487.
Acknowledgements
We thank the patients and their families for their participation in research studies. We are also thankful to Drs William Rhead, Peter Francis, Omar Abdul-Rahman, and Annet Van Hagen for referral of patients; Jennifer Rigdon, Kate Durda, Jamie L'Heureux, and Ecaterina Dragan for assistance with sample processing and clinical data collection; and Rachel Lorier, Stephen Hall, Katie Felhofer and Andrea Lenarduzzie for assistance with Affymetrix array CNV analysis. This work was supported by the National Institutes of Health awards EY015518 (EVS), 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) Program, Research Training Program in Vision Sciences T32 EY014537, and the Foerderer Fund (AVL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Reis, L., Tyler, R., Volkmann Kloss, B. et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet 20, 1224–1233 (2012). https://doi.org/10.1038/ejhg.2012.80
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2012.80
Keywords
This article is cited by
-
CRISPR-Cas9-mediated functional dissection of the foxc1 genomic region in zebrafish identifies critical conserved cis-regulatory elements
Human Genomics (2022)
-
A novel missense mutation of FOXC1 in an Axenfeld–Rieger syndrome patient with a congenital atrial septal defect and sublingual cyst: a case report and literature review
BMC Medical Genomics (2021)
-
A novel variant in FOXC1 associated with atypical Axenfeld-Rieger syndrome
BMC Medical Genomics (2021)
-
A missense mutation in Pitx2 leads to early-onset glaucoma via NRF2-YAP1 axis
Cell Death & Disease (2021)
-
Pitx genes in development and disease
Cellular and Molecular Life Sciences (2021)
