
Abstract
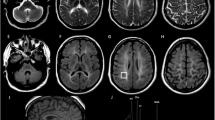
Homozygous or compound heterozygous mutations in the GJC2 gene, encoding the gap junction protein connexin47 (Cx47), cause the autosomal recessive hypomyelinating Pelizaeus–Merzbacher-like disease (PMLD1, MIM# 608804). Although clinical and neuroradiological findings resemble those of the classic Pelizaeus–Merzbacher disease, PMLD patients usually show a greater level of cognitive and motor functions. Unpredictably a homozygous missense GJC2 mutation (p.Glu260Lys) was found in a patient presenting with a very severe clinical picture characterised by congenital nystagmus and severe neurological impairment. Also magnetic resonance imaging was unusually severe, showing an abnormal supra- and infratentorial white matter involvement extending to the spinal cord. The novel p.Glu260Lys (c.778G>A) mutation, occurring in a highly conserved motif (SRPTEK) of the Cx47 extracellular loop-2 domain, was predicted, by modelling analysis, to break a ‘salt bridge network’, crucial for a proper connexin–connexin interaction to form a connexon, thus hampering the correct formation of the connexon pore. The same structural analysis, extended to the previously reported missense mutations, predicted that most changes were expected to have less severe impact on protein functions, correlating with the mild PMLD1 form of the patients. Our study expands the spectrum of PMLD1 and provides evidence that the extremely severe clinical and neuroradiological PMLD1 form of our patient likely correlates with the predicted impairment of gap junction channel assembly resulting from the detrimental effect of the new p.Glu260Lys mutant allele on Cx47 protein.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Accession codes
References
Garbern JY, Hobson GH PLP1-Related Disorders. GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle 1993 http://www.genetests.org (Updated 16 March 2010).
Uhlenberg B, Schuelke M, Ruschendorf F et al. Mutations in the gene encoding gap junction protein α12 (Connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet 2004; 75: 251–260.
Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS : Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia 2004; 47: 346–357.
Bugiani M, Al Shahwan S, Lamantea E et al. GJA12 mutations in children with recessive hypomielinating leukoencephalopathy. Neurology 2006; 67: 273–279.
Salviati L, Trevisson E, Baldoin MC et al. A novel deletion in the GJA12 gene causes Pelizaeus-Merzbacher-like disease. Neurogenetics 2007; 8: 57–60.
Wolf NI, Cundall M, Rutland P et al. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination. Neurogenetics 2007; 8: 39–44.
Henneke M, Combes P, Diekmann S et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology 2008; 70: 748–754.
Sartori S, Burlina AB, Salviati L et al. Increased level of N-acetylaspartylglutamate (NAAG) in the CSF of a patient with Pelizaeus-Merzbacher-like disease due to mutation in the GJA12 gene. Eur J Paediatr Neurol 2008; 12: 348–350.
Wang J, Wang H, Wang Y, Chen T, Wu X, Jiang Y : Two novel gap junction protein alpha 12 gene mutations in two Chinese patients with Pelizaeus-Merzbacher-like disease. Brain Dev 2010; 32: 236–243.
Osaka H, Hamanoue H, Yamamoto R et al. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease. Ann Neurol 2010; 68: 250–254.
Combes P, Kammoun N, Monnier A et al. Relevance of GJC2 promoter mutation in Pelizaeus-Merzbacher-like disease. Ann Neurol 2012; 71: 146–148.
Henneke M, Gegner S, Hahn A et al. Clinical neurophysiology in GJA12-related hypomyelination vs Pelizaeus-Merzbacher disease. Neurology 2010; 74: 1785–1789.
Meyer E, Kurian MA, Morgan NV et al. Promoter mutation is a common variant in GJC2-associated Pelizaeus-Merzbacher-like disease. Mol Genet Metab 2011; 104: 637–643.
Orthmann-Murphy JL, Salsano E, Abrams CK et al. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 2009; 132: 426–438.
Cailloux F, Gauthier-Barichard F, Mimault C et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet 2000; 8: 837–845.
Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS : Loss-of –function connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci 2007; 34: 629–641.
Sali A, Blundell TL : Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234: 779–815.
Maeda S, Nakagawa S, Suga M et al. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 2009; 458: 597–602.
Emsley P, Cowtan K : Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60 (Pt 12 Pt 1): 2126–2132.
Pettersen EF, Goddard TD, Huang CC et al. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612.
Ruf N, Uhlenberg B : Analysis of human alternative first exons and copy number variation of the GJA12 gene in patients with Pelizaeus-Merzbacher-like disease. Am J Med Genet Part B 2008; 150B: 226–232.
Steenweg ME, Vanderver A, Blaser S et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010; 133: 2971–2982.
Kovacs JA, Baker KA, Altenberg GA, Abagyan R, Yeager M : Molecular modeling and mutagenesis of gap junction channels. Prog Biophys Mol Biol 2007; 94: 15–28.
Biebermann H, Schoneberg T, Schulz A et al. A conserved tyrosine residue (Y601) in transmembrane domain 5 of the human thyrotropin receptor serves as a molecular switch to determine G-protein coupling. FASEB J 1998; 12: 1461–1471.
Gazit E : A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J 2002; 16: 77–83.
Sal-Man N, Gerber D, Bloch I, Shai Y : Specificity in transmembrane helix-helix interactions mediated by aromatic residues. J Biol Chem 2007; 282: 19753–19761.
The UniProt Consortium Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res 2011; 39: D214–D219.
Acknowledgements
The patient sample was obtained from the ‘Cell Line and DNA Biobank from Patients Affected by Genetic Diseases’ (G Gaslini Institute)—Telethon Genetic Biobank Network (Project No. GTB07001). The study was also supported by Italian Health Department ‘Finanziamento Ricerca Corrente (contributo per la ricerca intramurale)’ and FP7-HEALTH—LeukoTreat no.241622.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Biancheri, R., Rosano, C., Denegri, L. et al. Expanded spectrum of Pelizaeus–Merzbacher-like disease: literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet 21, 34–39 (2013). https://doi.org/10.1038/ejhg.2012.93
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2012.93
Keywords
This article is cited by
-
Novel mutations in the GJC2 gene associated with Pelizaeus–Merzbacher-like disease
Molecular Biology Reports (2019)
-
Mechanisms linking connexin mutations to human diseases
Cell and Tissue Research (2015)
