
Abstract
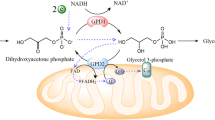
The constellation of clinico-pathological and laboratory findings including massive hepatomegaly, steatosis, and marked hypertriglyceridemia in infancy is extremely rare. We describe a child who is presented with the above findings, and despite extensive diagnostic testing no cause could be identified. Whole exome sequencing was performed on the patient and parents’ DNA. Mutations in GPD1 encoding glycerol-3-phosphate dehydrogenase that catalyzes the reversible redox reaction of dihydroxyacetone phosphate and NADH to glycerol-3-phosphate (G3P) and NAD+ were identified. The proband inherited a GPD1 deletion from the father determined using copy number analysis and a missense change p.(R229Q) from the mother. GPD1 protein was absent in the patient’s liver biopsy on western blot. Low normal activity of carnitine palmitoyl transferases, CPT1 and CPT2, was present in the patient’s skin fibroblasts, without mutations in genes encoding for these proteins. This is the first report of compound heterozygous mutations in GPD1 associated with a lack of GPD1 protein and reduction in CPT1 and CPT2 activity.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Menaya J, Gonzalez-Manchon C, Parrilla R, Ayuso MS : Molecular cloning, sequencing and expression of a cDNA encoding a human liver NAD-dependent alpha-glycerol-3-phosphate dehydrogenase. Biochim Biophys Acta 1995; 1262: 91–94.
Basel-Vanagaite L, Zevit N, Har Zahav A et al: Transient infantile hypertriglyceridemia, fatty liver, and hepatic fibrosis caused by mutated GPD1, encoding glycerol-3-phosphate dehydrogenase 1. Am J Hum Genet 2012; 90: 49–60.
Peterfy M, Ben-Zeev O, Mao HZ et al: Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat Genet 2007; 39: 1483–1487.
Brahm A, Hegele RA : Hypertriglyceridemia. Nutrients 2013; 5: 981–1001.
Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J : Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 2004; 25: 495–520.
Mathur A, Sims HF, Gopalakrishnan D et al: Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999; 99: 1337–1343.
Cakir M, Bruno C, Cansu A, Cobanoglu U, Erduran E : Liver cirrhosis in an infant with Chanarin-Dorfman syndrome caused by a novel splice-site mutation in ABHD5. Acta Paediatr 2010; 99: 1592–1594.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Ng PC, Henikoff S : SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814.
MacDonald MJ, Marshall LK : Mouse lacking NAD+-linked glycerol phosphate dehydrogenase has normal pancreatic beta cell function but abnormal metabolite pattern in skeletal muscle. Arch Biochem Biophys 2000; 384: 143–153.
Hajra AK, Bishop JE : Glycerolipid biosynthesis in peroxisomes via the acyl dihydroxyacetone phosphate pathway. Ann N Y Acad Sci 1982; 386: 170–182.
Agranoff BW, Hajra AK : The acyl dihydroxyacetone phosphate pathway for glycerolipid biosynthesis in mouse liver and Ehrlich ascites tumor cells. Proc Natl Acad Sci USA 1971; 68: 411–415.
Saggerson D : Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr 2008; 28: 253–272.
Acknowledgements
We acknowledge the financial support from the Gene Discovery Core of The Manton Center for Orphan Disease Research at Boston Children’s Hospital. Sanger sequencing was performed by the Molecular Genetics Core Facility of the IDDRC at Boston Children's Hospital supported by National Institute of Health (NIH) P30HD18655. PBA was supported by K08 AR055072 from the NIH.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Joshi, M., Eagan, J., Desai, N. et al. A compound heterozygous mutation in GPD1 causes hepatomegaly, steatohepatitis, and hypertriglyceridemia. Eur J Hum Genet 22, 1229–1232 (2014). https://doi.org/10.1038/ejhg.2014.8
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2014.8
Keywords
This article is cited by
-
Allosteric activation of the metabolic enzyme GPD1 inhibits bladder cancer growth via the lysoPC-PAFR-TRPV2 axis
Journal of Hematology & Oncology (2022)
-
Prognostic value of a novel glycolysis-related gene expression signature for gastrointestinal cancer in the Asian population
Cancer Cell International (2021)
-
Transient Infantile Hypertriglyceridemia and Hepatic Steatosis in an Infant with GPD1 Mutation
Indian Journal of Pediatrics (2021)
-
GPD1 Deficiency – Underdiagnosed Cause of Liver Disease
The Indian Journal of Pediatrics (2021)
-
A novel homozygous mutation in the glycerol-3-phosphate dehydrogenase 1 gene in a Chinese patient with transient infantile hypertriglyceridemia: a case report
BMC Gastroenterology (2018)
