
Abstract
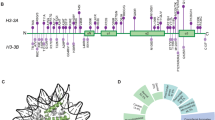
Truncating ASXL3 mutations were first identified in 2013 by Bainbridge et al. as a cause of syndromic intellectual disability in four children with similar phenotypes using whole-exome sequencing. The clinical features – postulated by Bainbridge et al. to be overlapping with Bohring–Opitz syndrome – were developmental delay, severe feeding difficulties, failure to thrive and neurological abnormalities. This condition was included in OMIM as ‘Bainbridge–Ropers syndrome’ (BRPS, #615485). To date, a total of nine individuals with BRPS have been published in the literature in four reports (Bainbridge et al., Dinwiddie et al, Srivastava et al. and Hori et al.). In this report, we describe six unrelated patients with newly diagnosed heterozygous de novo loss-of-function variants in ASXL3 and concordant clinical features: severe muscular hypotonia with feeding difficulties in infancy, significant motor delay, profound speech impairment, intellectual disability and a characteristic craniofacial phenotype (long face, arched eyebrows with mild synophrys, downslanting palpebral fissures, prominent columella, small alae nasi, high, narrow palate and relatively little facial expression). The majority of key features characteristic for Bohring–Opitz syndrome were absent in our patients (eg, the typical posture of arms, intrauterine growth retardation, microcephaly, trigonocephaly, typical facial gestalt with nevus flammeus of the forehead and exophthalmos). Therefore we emphasize that BRPS syndrome, caused by ASXL3 loss-of-function variants, is a clinically distinct intellectual disability syndrome with a recognizable phenotype distinguishable from that of Bohring–Opitz syndrome.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Kochinke K, Zweier C, Nijhof B et al: Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet 2016; 98: 149–164.
Bainbridge MN, Hu H, Muzny DM et al: De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med 2013; 5: 11.
Oberklaid F, Danks DM : The Opitz trigonocephaly syndrome. A case report. Am J Dis Child 1975; 129: 1348–1349.
Bohring A, Silengo M, Lerone M et al: Severe end of Opitz trigonocephaly (C) syndrome or new syndrome? Am J Med Genet 1999; 85: 438–446.
Hastings R, Cobben JM, Gillessen-Kaesbach G et al: Bohring-Opitz (Oberklaid-Danks) syndrome: clinical study, review of the literature, and discussion of possible pathogenesis. Eur J Hum Genet 2011; 19: 513–519.
Hoischen A, van Bon BWM, Rodríguez-Santiago B et al: De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet 2011; 43: 729–731.
Katoh M, Katoh M : Identification and characterization of ASXL3 gene in silico. Int J Oncol 2004; 24: 1617–1622.
Katoh M : Functional and cancer genomics of ASXL family members. Br J Cancer 2013; 109: 299–306.
Katoh M : Functional proteomics of the epigenetic regulators ASXL1, ASXL2 and ASXL3: a convergence of proteomics and epigenetics for translational medicine. Expert Rev Proteomics 2015; 12: 317–328.
Dinwiddie DL, Soden SE, Saunders CJ et al: De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC Med Genomics 2013; 6: 32.
Srivastava A, Ritesh KC, Tsan YC et al: De novo dominant ASXL3 mutations alter H2A deubiquitination and transcription in Bainbridge-Ropers syndrome. Hum Mol Genet 2016; 25: 597–608.
Hori I, Miya F, Ohashi K et al: Novel splicing mutation in the ASXL3 gene causing Bainbridge-Ropers syndrome. Am J Med Genet 2016; 170: 1863–1867.
Gillissen C, Hehir-Kwa JY, Thung DT et al: Genome sequencing identifies major causes of severe intellectual disability. Nature 2014; 511: 344–347.
Kuechler A, Zink AM, Wieland T et al: Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur J Hum Genet 2015; 23: 753–760.
Thevenon J, Duffourd Y, Masurel-Paulet A et al: Diagnostic odyssey in severe neurodevelopmental disorders: towards clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet 2016; 89: 700–707.
De Rubeis S, He X, Goldberg AP et al: Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215.
Russell B, Graham JM Jr : Expanding our knowledge of conditions associated with the ASXL gene family. Genome Med 2013; 5: 16.
Ropers HH, Wienker T : Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders. Eur J Med Genet 2015; 58: 715–718.
Arunachal G, Danda S, Omprakash S, Kumar S : A novel de-novo frameshift mutation of the ASXL1 gene in a classic case of Bohring-Opitz syndrome. Clin Dysmorphol 2016; 25: 101–105.
Huang N, Lee I, Marcotte EM, Hurles ME : Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 2010; 6: e1001154.
Hayden EC : Data barriers limit genetic diagnosis. Nature 2013; 494: 156–157.
Acknowledgements
We are grateful to the patients and their families for participating in this study and for giving consent to publish data and photographs. We thank Sabine Kaya and Daniela Falkenstein for excellent technical assistance. Parts of this study were supported by a grant from the Interdisciplinary Center for Clinical Research (laboratory rotation) of the Clinical Center Erlangen of the Friedrich-Alexander-Universität Erlangen-Nürnberg (to UH), the German Ministry of Research and Education (grant numbers 01GS08164, 01GS08167, 01GS08163, German Mental Retardation Network) as part of the National Genome Research Network (to HE), the German Network for Mitochondrial Disorders (mitoNET; 01GM1113 to HP), the E-Rare project GENOMIT (01GM1207 to HP) and the Juniorverbund in der Systemmedizin 'mitOmics' (FKZ 01ZX1405C to TBH). We thank the PARI 2012 Marfanoid habitus and intellectual deficiency (Regional Council of Burgundy and Dijon University Hospital, France) for its financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kuechler, A., Czeschik, J., Graf, E. et al. Bainbridge–Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition. Eur J Hum Genet 25, 183–191 (2017). https://doi.org/10.1038/ejhg.2016.165
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2016.165
This article is cited by
-
A case of Bainbridge-Ropers syndrome with breath holding spells and intractable epilepsy: challenges in diagnosis and management
BMC Neurology (2022)
-
Case report : a novel ASXL3 gene variant in a Sudanese boy
BMC Pediatrics (2021)
-
Emerging multifaceted roles of BAP1 complexes in biological processes
Cell Death Discovery (2021)
-
Compound heterozygous mutation of the ASXL3 gene causes autosomal recessive congenital heart disease
Human Genetics (2021)
-
Bainbridge-ropers syndrome caused by loss-of-function variants in ASXL3: Clinical abnormalities, medical imaging features, and gene variation in infancy of case report
BMC Pediatrics (2020)
