
Abstract
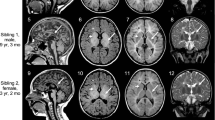
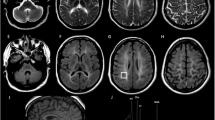
Pelizaeus–Merzbacher disease (PMD) is a rare Mendelian disorder characterised by central nervous system hypomyelination. PMD typically manifests in infancy or early childhood and is caused by mutations in proteolipid protein-1 (PLP1). However, variants in several other genes including gap junction protein gamma 2 (GJC2) can also cause a similar phenotype and are referred to PMD-like disease (PMLD). Whole-exome sequencing in two siblings presenting with clinical symptoms of PMD revealed a homozygous variant in the arginyl-tRNA synthetase (RARS) gene: NM_002887.3: c.[5A>G] p.(Asp2Gly). Subsequent screening of a PMD cohort without a genetic diagnosis identified an unrelated individual with novel compound heterozygous variants including a missense variant c.[1367C>T] p.(Ser456Leu) and a de novo deletion c.[1846_1847delTA] p.(Tyr616Leufs*6). Protein levels of RARS and the multi-tRNA synthetase complex into which it assembles were found to be significantly reduced by 80 and 90% by western blotting and Blue native-PAGE respectively using patient fibroblast extracts. As RARS is involved in protein synthesis whereby it attaches arginine to its cognate tRNA, patient cells were studied to determine their ability to proliferate with limiting amounts of this essential amino acid. Patient fibroblasts cultured in medium with limited arginine at 30 °C and 40 °C, showed a significant decrease in fibroblast proliferation (P<0.001) compared to control cells, suggestive of inefficiency of protein synthesis in the patient cells. Our functional studies provide further evidence that RARS is a PMD-causing gene.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Lossos A, Elazar N, Lerer I et al: Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder. Brain 2015; 138: 2521–2536.
Brender T, Wallerstein D, Sum J, Wallerstein R : Unusual presentation of Pelizaeus-Merzbacher disease: female patient with deletion of the proteolipid protein 1 gene. Case Rep Genet 2015; 2015: 453105.
Golomb MR, Walsh LE, Carvalho KS, Christensen CK, DeMyer WE : Clinical findings in Pelizaeus-Merzbacher disease. J Child Neurol 2004; 19: 328–331.
Sima AA, Pierson CR, Woltjer RL et al: Neuronal loss in Pelizaeus-Merzbacher disease differs in various mutations of the proteolipid protein 1. Acta Neuropathol 2009; 118: 531–539.
Xia J, Wang L : Pelizaeus-Merzbacher disease: molecular diagnosis and therapy. Intractable Rare Dis Res 2013; 2: 103–105.
Hobson GM, Kamholz J PLP1-related disorders;in Pagon RA, Adam MP, Ardinger HH et al(eds):GeneReviews. Seattle: University of Washington, 1993.
Carvalho CM, Bartnik M, Pehlivan D, Fang P, Shen J, Lupski JR : Evidence for disease penetrance relating to CNV size: Pelizaeus-Merzbacher disease and manifesting carriers with a familial 11 Mb duplication at Xq22. Clin Genet 2012; 81: 532–541.
Hobson GM, Garbern JY : Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol 2012; 32: 62–67.
Karimzadeh P, Ahmadabadi F, Aryani O, Houshmand M, Khatami A : New mutation of pelizaeus—merzbacher-like disease; a report from iran. Iranian J Radiol 2014; 11: e6913.
Feinstein M, Markus B, Noyman I et al: Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820–828.
Magen D, Georgopoulos C, Bross P et al: Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008; 83: 30–42.
Miyamoto Y, Torii T, Eguchi T, Nakamura K, Tanoue A, Yamauchi J : Hypomyelinating leukodystrophy-associated missense mutant of FAM126A/hyccin/DRCTNNB1A aggregates in the endoplasmic reticulum. J Clin Neurosci 2014; 21: 1033–1039.
Shimojima K, Okumura A, Ikeno M et al: A de novo TUBB4A mutation in a patient with hypomyelination mimicking Pelizaeus-Merzbacher disease. Brain Dev 2015; 37: 281–285.
Wolf NI, Vanderver A, van Spaendonk RM et al: Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014; 83: 1898–1905.
Wolf NI, Salomons GS, Rodenburg RJ et al: Mutations in RARS cause hypomyelination. Ann Neurol 2014; 76: 134–139.
Nakayama T, Al-Maawali A, El-Quessny M et al: Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am J Hum Genet 2015; 96: 709–719.
Edvardson S, Gerhard F, Jalas C et al: Hypomyelination and developmental delay associated with VPS11 mutation in Ashkenazi-Jewish patients. J Med Genet 2015; 52: 749–753.
Thiffault I, Wolf NI, Forget D et al: Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015; 6: 7623.
Lopez-Espindola D, Morales-Bastos C, Grijota-Martinez C et al: Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab 2014; 99: E2799–E2804.
Cayami FK, La Piana R, van Spaendonk RM et al: POLR3A and POLR3B mutations in unclassified hypomyelination. Neuropediatrics 2015; 46: 221–228.
Biancheri R, Rossi A, Zara F, Filocamo M : AIMP1/p43 mutation and PMLD. Am J Hum Genet 2011; 88: 391, author reply 393-395.
Eriani G, Delarue M, Poch O, Gangloff J, Moras D : Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 1990; 347: 203–206.
Delarue M, Moras D : The aminoacyl-tRNA synthetase family: modules at work. BioEssays 1993; 15: 675–687.
Rémion A, Khoder‐Agha F, Cornu D, Argentini M, Redeker V, Mirande M : Identification of protein interfaces within the multi-aminoacyl-tRNA synthetase complex: the case of lysyl-tRNA synthetase and the scaffold protein p38. FEBS Open Bio 2016; 6: 696–706.
Li R, Macnamara LM, Leuchter JD, Alexander RW, Cho SS : MD Simulations of tRNA and Aminoacyl-tRNA synthetases: dynamics, folding, binding, and allostery. Intern J Molec Sci 2015; 16: 15872–15902.
Park SJ, Ahn HS, Kim JS, Lee C : Evaluation of Multi-tRNA synthetase complex by multiple reaction monitoring mass spectrometry coupled with size exclusion chromatography. PLoS One 2015; 10: e0142253.
Kim HS, Cha SY, Jo CH, Han A, Hwang KY : The crystal structure of arginyl-tRNA synthetase from Homo sapiens. FEBS Lett 2014; 588: 2328–2334.
Yang F, Ji QQ, Ruan LL, Ye Q, Wang ED : The mRNA of human cytoplasmic arginyl-tRNA synthetase recruits prokaryotic ribosomes independently. J Biol Chem 2014; 289: 20953–20959.
Havrylenko S, Mirande M : Aminoacyl-tRNA synthetase complexes in evolution. Int J Mol Sci 2015; 16: 6571–6594.
Zheng YG, Wei H, Ling C, Xu MG, Wang ED : Two forms of human cytoplasmic arginyl-tRNA synthetase produced from two translation initiations by a single mRNA. Biochemistry 2006; 45: 1338–1344.
Alodaib A, Sobreira N, Gold WA et al: Whole-exome sequencing identifies novel variants in PNPT1 causing oxidative phosphorylation defects and severe multisystem disease. Eur J Hum Genet 2016; 25: 79–84.
Riley LG, Menezes MJ, Rudinger-Thirion J et al: Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J Rare Dis 2013; 8: 193.
Guo M, Schimmel P, Yang XL : Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett 2010; 584: 434–442.
Pang YLJ, Poruri K, Martinis SA : tRNA synthetase: tRNA Aminoacylation and beyond. Wiley Interdiscip Rev RNA 2014; 5: 461–480.
Kyriacou SV, Deutscher MP : An important role for the multienzyme aminoacyl-tRNA synthetase complex in mammalian translation and cell growth. Mol Cell 2008; 29: 419–427.
Venselaar H, te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G : Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11: 548–548.
Arnez JG, Moras D : Structural and functional considerations of the aminoacylation reaction. Trends Biochem Sci 1997; 22: 211–216.
Popp MW, Maquat LE : Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell 2016; 165: 1319–1322.
Simons C, Griffin LB, Helman G et al: Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet 2015; 96: 675–681.
Szpisjak L, Zsindely N, Engelhardt JI, Vecsei L, Kovacs GG, Klivenyi P : Novel AARS2 gene mutation producing leukodystrophy: a case report. J Hum Genet 2017; 62: 329–333.
Wang J-C, Ross L, Mahon LW et al: Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur J Hum Genet 2015; 23: 663–671.
Wang K, Kim C, Bradfield J et al: Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med 2013; 5: 67.
Niehues S, Bussmann J, Steffes G et al: Impaired protein translation in drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat Commun 2015; 6: 7520.
Nangle LA, Zhang W, Xie W, Yang XL, Schimmel P : Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci USA 2007; 104: 11239–11244.
Acknowledgements
We are grateful to the NSW Biochemical Genetics Service for establishing initial fibroblast line cultures, and the Molecular Genetics Department for initial DNA extraction, both located at the Western Sydney Genetics Program, Children’s Hospital at Westmead. We are also grateful to Dr Grace Hobson, Alfred I DuPont Hospital for Children, Wilmington, DE, USA, for provision of DNA samples for Sanger sequencing, none of which were shown to have RARS variations. Finally, we are grateful to Dr Shanti Balasubramaniam (Western Sydney Genetics Program, Children’s Hospital at Westmead) and Dr Sebastian Lunke (Translational Genomics Unit, Victorian Clinical Genetics Services) for acquisition and interpretation of some data relating to patient 1. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients included in the study as approved by the Human Research Ethics Committee of the Sydney Children’s Hospitals Network and the Institutional Review Board of Johns Hopkins Medicine.
Author contributions
MN: design and implementation of structural and functional studies, data analysis and manuscript preparation. WG and LR: contribution to design of structural and functional studies, data analysis and manuscript preparation. NS and CB: performance of whole-exome sequencing, implementation of bioinformatics, data analysis and manuscript preparation. KP: interpretation of MRI images for the Australian patients and manuscript preparation. BU: Initial molecular genetic screening and provision DNA sample for the German patient and manuscript preparation. CW: provision of clinical information for the German patient and manuscript preparation. RO: clinical diagnosis and management of the Australian patients and manuscript preparation. JC: overall oversight of the research project, clinical interface, data analysis and manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nafisinia, M., Sobreira, N., Riley, L. et al. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus–Merzbacher disease. Eur J Hum Genet 25, 1134–1141 (2017). https://doi.org/10.1038/ejhg.2017.119
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2017.119
This article is cited by
-
Uncovering potential causal genes for undiagnosed congenital anomalies using an in-house pipeline for trio-based whole-genome sequencing
Human Genomics (2025)
-
Homozygous EPRS1 missense variant causing hypomyelinating leukodystrophy-15 alters variant-distal mRNA m6A site accessibility
Nature Communications (2024)
-
A Cysteinyl-tRNA Synthetase Mutation Causes Novel Autosomal-Dominant Inheritance of a Parkinsonism/Spinocerebellar-Ataxia Complex
Neuroscience Bulletin (2024)
-
Four pedigrees with aminoacyl-tRNA synthetase abnormalities
Neurological Sciences (2022)
-
Knockdown of Golgi Stress-Responsive Caspase-2 Ameliorates HLD17-Associated AIMP2 Mutant-Mediated Inhibition of Oligodendroglial Cell Morphological Differentiation
Neurochemical Research (2022)
