
Abstract
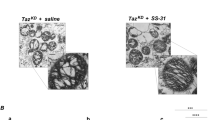
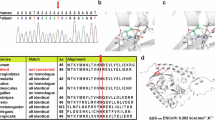
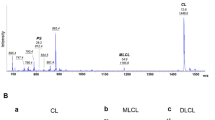
Barth syndrome (BTHS) is a rare X-linked disorder characterized by cardiomyopathy, short stature, neutropenia, and 3-methylglutaconic aciduria. Mutations have been identified in the TAZ (G4.5) gene in patients with BTHS. This article presents a mutation analysis of this gene in a Japanese boy with cardiomyopathy with abnormal mitochondria, cyclic neutropenia, and 3-methylglutaconic aciduria (type 2). The analysis revealed a novel missense mutation (R94S) caused by a single nucleotide substitution (C-to-A) in this patient.
Similar content being viewed by others
Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Author information
Authors and Affiliations
Additional information
Received: November 30, 2001 / Accepted: February 9, 2002
Rights and permissions
About this article
Cite this article
Sakamoto, O., Kitoh, T., Ohura, T. et al. Novel missense mutation (R94S) in the TAZ (G4.5) gene in a Japanese patient with Barth syndrome. J Hum Genet 47, 229–231 (2002). https://doi.org/10.1007/s100380200030
Issue date:
DOI: https://doi.org/10.1007/s100380200030
This article is cited by
-
Mis-sesnse mutations in Tafazzin (TAZ) that escort to mild clinical symptoms of Barth syndrome is owed to the minimal inhibitory effect of the mutations on the enzyme function: In-silico evidence
Interdisciplinary Sciences: Computational Life Sciences (2015)
-
Natural history of Barth syndrome: a national cohort study of 22 patients
Orphanet Journal of Rare Diseases (2013)
