
Abstract
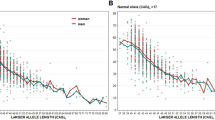
We studied the allelic profile of CAG and CCG repeats in 61 Brazilian individuals in 21 independent families affected by Huntington’s disease (HD). Thirteen individuals had two normal alleles for HD, two had one mutable normal allele and no HD phenotype, and forty-six patients carried at least one expanded CAG repeat allele. Forty-five of these individuals had one expanded allele and one individual had one mutable normal allele (27 CAG repeats) and one expanded allele (48 CAG repeats). Eleven of these forty-five subjects had a mutant allele with reduced penetrance, and thirty-four patients had a mutant allele with complete penetrance. Inter- and intragenerational investigations of CAG repeats were also performed. We found a negative correlation between the number of CAG repeats and the age of disease onset (r=−0.84; P<0.001) and no correlation between the number of CCG repeats and the age of disease onset (r=0.06). We found 40 different haplotypes and the analysis showed that (CCG)10 was linked to a CAG normal allele in 19 haplotypes and to expanded alleles in two haplotypes. We found that (CCG)7 was linked to expanded CAG repeats in 40 haplotypes (95.24%) and (CCG)10 was linked to expanded CAG repeats in only two haplotypes (4.76%). Therefore, (CCG)7 was the most common allele in HD chromosomes in this Brazilian sample. It was also observed that there was a significant association of (CCG)7 with the expanded CAG alleles (χ2=6.97, P=0.0084). Worldwide, the most common CCG alleles have 7 or 10 repeats. In Western Europe, (CCG)7 is the most frequent allele, similarly to our findings.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Change history
21 December 2012
This article has been corrected since Advance Online Publication, and an erratum is also printed in this issue.
References
The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983 (1993).
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L. et al. Loss of Huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293, 493–498 (2001).
Andrew, S. E., Goldberg, Y. P., Kremer, B., Telenius, H., Theilmann, J., Adam, S. et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 4, 398–403 (1993).
Nahhas, F. A., Garbern, J., Krajewski, K. M., Roa, B. B. & Feldman, G. L. Juvenile onset Huntington disease resulting from a very large maternal expansion. Am. J. Med. Genet. A 137A, 328–331 (2005).
Beighton, P. & Hayden, M. R. Huntington’s chorea. S. Afr. Med. J. 59, 250 (1981).
Hayden, M. R., Goldblatt, J., Wallis, G., Winship, I. M. & Beighton, P. Molecular genetics and Huntington’s disease. The South African situation. S. Afr. Med. J 71, 683–686 (1987).
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P. et al. Aggregation of Huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 (1997).
Gil, J. M. & Rego, A. C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci 27, 2803–2820 (2008).
Bonelli, R. M. & Hofmann, P. A systematic review of the treatment studies in Huntington's disease since 1990. Expert Opin. Pharmacother. 8, 141–153 (2007).
ACMG/ASHG Statement. Laboratory guidelines for Huntington disease genetic testing. The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group. Am. J. Hum. Genet. 62, 1243–1247 (1998).
Woolf, B. On estimating the relation between blood group and disease. Ann. Hum. Genet. 19, 251–253 (1955).
Haldane, J. B. S. & Smith, SM. The sampling distribution of a maximum likelihood estimate. Biometrika 43, 96–103 (1956).
Lima, E. S. T. C., Serra, H. G., Bertuzzo, C. S. & Lopes-Cendes, I. Molecular diagnosis of Huntington disease in Brazilian patients. Arq Neuropsiquiatr. 58, 11–17 (2000).
Raskin, S., Allan, N., Teive, H. A., Cardoso, F., Haddad, M. S., Levi, G. et al. Huntington disease: DNA analysis in Brazilian population. Arq Neuropsiquiatr. 58, 977–985 (2000).
Morovvati, S., Nakagawa, M., Osame, M. & Karami, A. Analysis of CCG repeats in Huntingtin gene among HD patients and normal populations in Japan. Arch. Med. Res. 39, 131–133 (2008).
Andrich, J., Arning, L., Wieczorek, S., Kraus, P. H., Gold, R. & Saft, C. Huntington’s disease as caused by 34 CAG repeats. Mov. Disord. 23, 879–881 (2008).
Telenius, H., Kremer, B., Goldberg, Y. P., Theilmann, J., Andrew, S. E., Zeisler, J. et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet. 6, 409–414 (1994).
Seneca, S., Fagnart, D., Keymolen, K., Lissens, W., Hasaerts, D., Debulpaep, S. et al. Early onset Huntington disease: a neuronal degeneration syndrome. Eur. J. Pediatr. 163, 717–721 (2004).
Nance, M. A., Mathias-Hagen, V., Breningstall, G., Wick, M. J. & McGlennen, R. C. Analysis of a very large trinucleotide repeat in a patient with juvenile Huntington’s disease. Neurology 52, 392–394 (1999).
Wexler, N. S., Lorimer, J., Porter, J., Gomez, F., Moskowitz, C., Shackell, E. et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl Acad. Sci. USA 101, 3498–3503 (2004).
Muglia, M., Leone, O., Annesi, G., Gabriele, A. L., Imbrogno, E., Grandinetti, C. et al. Nonisotopic method for accurate detection of (CAG)n repeats causing Huntington disease. Clin. Chem. 42, 1601–1603 (1996).
Squitieri, F., Andrew, S. E., Goldberg, Y. P., Kremer, B., Spence, N., Zeisler, J. et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum. Mol. Genet. 3, 2103–2114 (1994).
Pramanik, S., Basu, P., Gangopadhaya, P. K., Sinha, K. K., Jha, D. K., Sinha, S. et al. Analysis of CAG and CCG repeats in Huntingtin gene among HD patients and normal populations of India. Eur. J. Hum. Genet. 8, 678–682 (2000).
Hecimovic, S., Klepac, N., Vlasic, J., Vojta, A., Janko, D., Skarpa-Prpic, I. et al. Genetic background of Huntington disease in Croatia: molecular analysis of CAG, CCG, and Delta2642 (E2642del) polymorphisms. Hum. Mutat. 20, 233 (2002).
Panegyres, P. K., Beilby, J., Bulsara, M., Toufexis, K. & Wong, C. A study of potential interactive genetic factors in Huntington’s disease. Eur. Neurol. 55, 189–192 (2006).
Chattopadhyay, B., Ghosh, S., Gangopadhyay, P. K., Das, S. K., Roy, T., Sinha, K. K. et al. Modulation of age at onset in Huntington’s disease and spinocerebellar ataxia type 2 patients originated from eastern India. Neurosci. Lett. 345, 93–96 (2003).
Costa, M. C., Magalhaes, P., Guimaraes, L., Maciel, P., Sequeiros, J. & Sousa, A. The CAG repeat at the Huntington disease gene in the Portuguese population: insights into its dynamics and to the origin of the mutation. J. Hum. Genet. 51, 189–195 (2006).
Masuda, N., Goto, J., Murayama, N., Watanabe, M., Kondo, I. & Kanazawa, I. Analysis of triplet repeats in the Huntingtin gene in Japanese families affected with Huntington’s disease. J. Med. Genet. 32, 701–705 (1995).
Wang, C. K., Wu, Y. R., Hwu, W. L., Chen, C. M., Ro, L. S., Chen, S. T. et al. DNA haplotype analysis of CAG repeat in Taiwanese Huntington’s disease patients. Eur. Neurol. 52, 96–100 (2004).
Rubinsztein, D. C., Amos, W., Leggo, J., Goodburn, S., Ramesar, R. S., Old, J. et al. Mutational bias provides a model for the evolution of Huntington’s disease and predicts a general increase in disease prevalence. Nat. Genet. 7, 525–530 (1994).
Wheeler, V. C., Persichetti, F., McNeil, S. M., Mysore, J. S., Mysore, S. S., MacDonald, M. E. et al. Factors associated with HD CAG repeat instability in Huntington disease. J. Med. Genet. 44, 695–701 (2007).
Kovtun, I. V., Spiro, C. & McMurray, C. T. Triplet repeats and DNA repair: germ cell and somatic cell instability in transgenic mice. Methods Mol. Biol. 277, 309–319 (2004).
Hayden, M. R., Berkowicz, A. L., Beighton, P. H. & Yiptong, C. Huntington’s chorea on the island of Mauritius. S. Afr. Med. J 60, 1001–1002 (1981).
Garcia-Planells, J., Burguera, J. A., Solis, P., Millan, J. M., Ginestar, D., Palau, F. et al. Ancient origin of the CAG expansion causing Huntington disease in a Spanish population. Hum. Mutat. 25, 453–459 (2005).
Andrew, S. E. & Hayden, M. R. Origins and evolution of Huntington disease chromosomes. Neurodegeneration 4, 239–244 (1995).
Acknowledgements
Many thanks are due to Professor LCS Thuler and G Tucher for helping with data analysis. We are grateful to CAPES/PROAP/PPGNEURO, FINEP, UNIRIO, CNPq and FAPERJ for the financial support, and to FAMINAS, Associação Brasil Huntington (ABH) and the Department of Genetics and Molecular Biology for referring cases to the clinical geneticists.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Agostinho, L., Rocha, C., Medina-Acosta, E. et al. Haplotype analysis of the CAG and CCG repeats in 21 Brazilian families with Huntington’s disease. J Hum Genet 57, 796–803 (2012). https://doi.org/10.1038/jhg.2012.120
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2012.120
Keywords
This article is cited by
-
Investigation of the Influence of TBP CAG/CAA Repeats in Conjunction with HTT CAG Repeats on Huntington’s Disease Age at Onset in a Brazilian Sample
Journal of Molecular Neuroscience (2022)
-
Huntington’s Disease: Relationship Between Phenotype and Genotype
Molecular Neurobiology (2017)
