
Abstract
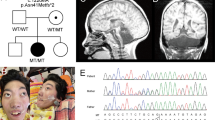
Pontocerebellar hypoplasia (PCH) is characterized by hypoplasia and atrophy of the cerebellum, variable pontine atrophy, microcephaly, severe mental and motor impairments and seizures. Mutations in 11 genes have been reported in 8 out of 10 forms of PCH. Recessive mutations in the mitochondrial arginyl-transfer RNA synthetase gene (RARS2) have been recently associated with PCH type 6, which is characterized by early-onset encephalopathy with signs of oxidative phosphorylation defect. Here we describe the clinical presentation, neuroimaging findings and molecular characterizations of two siblings with a clinical diagnosis of PCH who displayed a novel variant (c.-2A>G) in the 5′-UTR of the RARS2 gene in the homozygous state. This variant was identified through next-generation sequencing testing of a panel of nine genes known to be involved in PCH. Gene expression and functional studies demonstrated that the c.-2A>G sequence change directly leads to a reduced RARS2 messenger RNA expression in the patients by decreasing RARS2 promoter activity, thus providing evidence that mutations in the RARS2 promoter are likely to represent a new causal mechanism of PCH6.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Dyment, D. A., Sawyer, S. L., Chardon, J. W. & Boycott, K. M. Recent advances in the genetic etiology of brain malformations. Curr. Neurol. Neurosci. Rep. 13, 364 (2013).
Renbaum, P., Kellerman, E., Jaron, R., Geiger, D., Segel, R. & Lee, M. et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am. J. Hum. Genet. 85, 281–289 (2009).
Wan, J., Yourshaw, M., Mamsa, H., Rudnik-Schoneborn, S., Menezes, M. P. & Hong, J. E. et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat. Genet. 44, 704–708 (2012).
Barth, P. G. Pontocerebellar hypoplasia—how many types? Eur. J. Paediatr. Neurol. 4, 161–162 (2000).
Agamy, O., Ben Zeev, B., Lev, D., Marcus, B., Fine, D. & Su, D. et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Hum. Genet. 87, 538–544 (2010).
Budde, B. S., Namavar, Y., Barth, P. G., Poll-The, B. T., Nurnberg, G. & Becker, C. et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 40, 1113–1118 (2008).
Ben-Zeev, B., Hoffman, C., Lev, D., Watemberg, N., Malinger, G. & Brand, N. et al. Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J. Med. Genet. 40, e96 (2003).
Barth, P. G., Blennow, G., Lenard, H. G., Begeer, J. H., van der Kley, J. M. & Hanefeld, F. et al. The syndrome of autosomal recessive pontocerebellar hypoplasia, microcephaly, and extrapyramidal dyskinesia (pontocerebellar hypoplasia type 2): compiled data from 10 pedigrees. Neurology 45, 311–317 (1995).
Nakamura, K., Nishiyama, K., Kodera, H., Nakashima, M., Tsurusaki, Y. & Miyake, N. et al. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 36, 272–273 (2014).
Edvardson, S., Shaag, A., Kolesnikova, O., Gomori, J. M., Tarassov, I. & Einbinder, T. et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 81, 857–862 (2007).
Mochida, G. H., Ganesh, V. S., de Michelena, M. I., Dias, H., Atabay, K. D. & Kathrein, K. L. et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat. Genet. 44, 1260–1264 (2012).
Akizu, N., Cantagrel, V., Schroth, J., Cai, N., Vaux, K. & McCloskey, D. et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 154, 505–517 (2013).
Schaffer, A. E., Eggens, V. R., Caglayan, A. O., Reuter, M. S., Scott, E. & Coufal, N. G. et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157, 651–663 (2014).
Karaca, E., Weitzer, S., Pehlivan, D., Shiraishi, H., Gogakos, T. & Hanada, T. et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157, 636–650 (2014).
Cassandrini, D., Cilio, M. R., Bianchi, M., Doimo, M., Balestri, M. & Tessa, A. et al. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J. Inherit. Metab. Dis. 36, 43–53 (2013).
Rankin, J., Brown, R., Dobyns, W. B., Harington, J., Patel, J. & Quinn, M. et al. Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am. J. Med. Genet. A 152A, 2079–2084 (2010).
Namavar, Y., Barth, P. G., Kasher, P. R., van Ruissen, F., Brockmann, K. & Bernert, G. et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134, 143–156 (2011).
Glamuzina, E., Brown, R., Hogarth, K., Saunders, D., Russell-Eggitt, I. & Pitt, M. et al. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J. Inherit. Metab. Dis. 35, 459–467 (2012).
Cooper, D. N. Human gene mutation in pathology and evolution. J. Inherit. Metab. Dis. 25, 157–182 (2002).
de Vooght, K. M., van Wijk, R. & van Solinge, W. W. Management of gene promoter mutations in molecular diagnostics. Clin. Chem. 55, 698–708 (2009).
Deaton, A. M. & Bird, A. CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011).
Tie, F., Banerjee, R., Stratton, C. A., Prasad-Sinha, J., Stepanik, V. & Zlobin, A. et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141 (2009).
Pasini, D., Malatesta, M., Jung, H. R., Walfridsson, J., Willer, A. & Olsson, L. et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38, 4958–4969 (2010).
Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148 (1987).
Nakagawa, S., Niimura, Y., Gojobori, T., Tanaka, H. & Miura, K. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 36, 861–871 (2008).
Wolf, A., Caliebe, A., Thomas, N. S., Ball, E. V., Mort, M. & Stenson, P. D. et al. Single base-pair substitutions at the translation initiation sites of human genes as a cause of inherited disease. Hum. Mutat. 32, 1137–1143 (2011).
Kline-Fath B., R.B.-S., Bulas D. Fundamental and Advanced Fetal Imaging: Ultrasound and MRI, (Wolters Kluwer, Chicago, IL, USA, 2015).
Acknowledgements
We would like to thank the family for their participation in our study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, Z., Schonberg, R., Guidugli, L. et al. A novel mutation in the promoter of RARS2 causes pontocerebellar hypoplasia in two siblings. J Hum Genet 60, 363–369 (2015). https://doi.org/10.1038/jhg.2015.31
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2015.31
This article is cited by
-
A non-coding variant in the Kozak sequence of RARS2 strongly decreases protein levels and causes pontocerebellar hypoplasia
BMC Medical Genomics (2023)
-
What’s new in pontocerebellar hypoplasia? An update on genes and subtypes
Orphanet Journal of Rare Diseases (2018)
-
Novel homozygous RARS2 mutation in two siblings without pontocerebellar hypoplasia – further expansion of the phenotypic spectrum
Orphanet Journal of Rare Diseases (2016)
-
Structural basis for early-onset neurological disorders caused by mutations in human selenocysteine synthase
Scientific Reports (2016)
-
Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability
Metabolic Brain Disease (2016)
