
Abstract
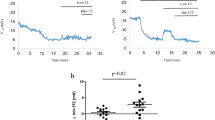
Cystic fibrosis (CF) is an autosomal recessive disease that results from mutations in the CF transmembrane conductance regulator (CFTR) gene. The effect of interventions aimed at correcting the CF electrophysiologic phenotype has been primarily measured using in vitro methods in gastrointestinal and respiratory epithelia. A reliable in vivo assay of CFTR function would be of great value in the investigation of pharmacologic interventions for CF mouse models. We performed the in vivo rectal potential difference (RPD) assay on three different mouse models. We then compared the in vivo data with the results obtained using the in vitro Ussing chamber method. The results from the in vitro method correlated closely with the results acquired using the in vivo method and were reproducible. The data suggest that the in vivo RPD assay is a reliable assay of functional CFTR expression in CF mouse models.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Abbreviations
- Bl6:
-
Black 6
- CF:
-
cystic fibrosis
- CFTR:
-
cystic fibrosis transmembrane conductance regulator
- CFTR(−/+):
-
heterozygous for wild type CFTR and mutant CFTR
- CFTRinh-172:
-
a specific CFTR channel inhibitor
- DCPD:
-
distal colon potential difference
- ΔF508:
-
deletion of phenylalanine at the 508 position
- ΔPDfsk:
-
mean change in potential difference in response to forskolin
- KBR:
-
Kreb's bicarbonate Ringer's solution
- PD:
-
potential difference
- RPD:
-
rectal potential difference
- TMAO:
-
trimethylamine oxide
References
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui L 1989 Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073
Davis PB, Drumm M, Konstan MW 1996 Cystic fibrosis. Am J Respir Crit Care Med 154: 1229–1256
Sheppard DN, Welsh MJ Structure and function of the CFTR chloride channel. Physiol Rev 1999 79: S23–S45
Thomas PJ, Shenbagamurthi P, Sondek J, Hullihen JM, Pedersen PL 1992 The cystic fibrosis transmembrane conductance regulator. Effects of the most common cystic fibrosis-causing mutation on the secondary structure and stability of a synthetic peptide. J Biol Chem 267: 5727–5730
Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE 1990 Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834
Ward CL, Omura S, Kopito RR 1995 Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83: 121–127
Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ 1992 Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358: 761–764
Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, Collins FS 1991 Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science 254: 1797–1799
Rubenstein RC, Egan ME, Zeitlin PL 1997 In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 100: 2457–2465
Fischer H, Fukuda N, Barbry P, Illek B, Sartori C, Matthay MA 2001 Partial restoration of defective chloride conductance in DeltaF508 CF mice by trimethylamine oxide. Am J Physiol Lung Cell Mol Physiol 281: L52–L57
Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ 2004 Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304: 600–602
Illek B, Fischer H, Santos GF, Widdicombe JH, Machen TE, Reenstra WW 1995 cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am J Physiol 268: C886–C893
Illek B, Fischer H 1998 Flavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivo. Am J Physiol 275: L902–L910
Fischer H, Schwarzer C, Illek B 2004 Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci U S A 101: 3691–3696
Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E 2003 Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 349: 1433–1441
Knowles M, Gatzy J, Boucher R 1981 Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 305: 1489–1495
Knowles M, Gatzy J, Boucher R 1983 Relative ion permeability of normal and cystic fibrosis nasal epithelium. J Clin Invest 71: 1410–1417
Grubb BR 2002 Bioelectric measurement of CFTR function in mice. In: Skach WR (ed) Methods in Molecular Medicine. Humana Press, Inc., Totowa, NJ, pp 525–535
Wilschanski MA, Rozmahel R, Beharry S, Kent G, Li C, Tsui LC, Durie P, Bear CE 1996 In vivo measurements of ion transport in long-living CF mice. Biochem Biophys Res Commun 219: 753–759
Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Capecchi MR, Welsh MJ, Thomas KR 1995 A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest 96: 2051–2064
Koller BH, Kim HS, Latour AM, Brigman K, Boucher RC Jr, Scambler P, Wainwright B, Smithies O 1991 Toward an animal model of cystic fibrosis: targeted interruption of exon 10 of the cystic fibrosis transmembrane regulator gene in embryonic stem cells. Proc Natl Acad Sci U S A 88: 10730–10734
Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH 1992 An animal model for cystic fibrosis made by gene targeting. Science 257: 1083–1088
Clarke LL, Gawenis LR, Franklin CL, Harline MC 1996 Increased survival of CFTR knockout mice with an oral osmotic laxative. Lab Anim Sci 46: 612–618
Taddei A, Folli C, Zeggra-Moran O, Fanen P, Verkman AS, Gailetta LJ 2004 Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett 558: 52–56
Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ 1994 Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266: 107–109
Cuthbert AW, Halsted J, Ratcliff R, Colledge WH, Evans MJ 1995 The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol 482: 449–454
Acknowledgements
We would like to acknowledge Dr. Horst Fischer for his extremely helpful communications during the development of our in vivo rectal potential difference assay. We would also like to acknowledge and thank Jeffrey Wisner for his extraordinary technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by grants from the National Institutes of Health (DK53428, MEE) and the Cystic Fibrosis Foundation (EGAN04G0, MEE.). S.A.W. is supported by a training grant from the National Institutes of Health (T32HD07094).
Rights and permissions
About this article
Cite this article
Weiner, S., Caputo, C., Bruscia, E. et al. Rectal Potential Difference and the Functional Expression of CFTR in the Gastrointestinal Epithelia in Cystic Fibrosis Mouse Models. Pediatr Res 63, 73–78 (2008). https://doi.org/10.1203/PDR.0b013e31815b4bc6
Received:
Accepted:
Issue date:
DOI: https://doi.org/10.1203/PDR.0b013e31815b4bc6
This article is cited by
-
The Ussing chamber system for measuring intestinal permeability in health and disease
BMC Gastroenterology (2019)
-
Colonic potassium handling
Pflügers Archiv - European Journal of Physiology (2010)
