
Abstract
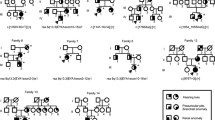
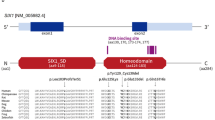
Branchio-oto-renal (BOR) syndrome is a rare autosomal dominant disorder characterized by branchiogenic anomalies, hearing loss, and renal anomalies. The aim of this study was to reveal the clinical phenotypes and their causative genes in Japanese BOR patients. Patients clinically diagnosed with BOR syndrome were analyzed by direct sequencing, multiplex ligation-dependent probe amplification (MLPA), array-based comparative genomic hybridization (aCGH), and next-generation sequencing (NGS). We identified the causative genes in 38/51 patients from 26/36 families; EYA1 aberrations were identified in 22 families, SALL1 mutations were identified in two families, and SIX1 mutations and a 22q partial tetrasomy were identified in one family each. All patients identified with causative genes suffered from hearing loss. Second branchial arch anomalies, including a cervical fistula or cyst, preauricular pits, and renal anomalies, were frequently identified (>60%) in patients with EYA1 aberrations. Renal hypodysplasia or unknown-cause renal insufficiency was identified in more than half of patients with EYA1 aberrations. Even within the same family, renal phenotypes often varied substantially. In addition to direct sequencing, MLPA and NGS were useful for the genetic analysis of BOR patients.
Similar content being viewed by others
Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Morisada N, Nozu K, Iijima K. Branchio-oto-renal syndrome: comprehensive review based on nationwide surveillance in Japan. Pediatr Int. 2014;56:309–14.
Smith RJH. In: Branchiootorenal spectrum disorders. Pagon RA, Adam MP, Ardinger HH, (eds). GeneReviews. Seattle: University of Washington; 2015.
Fraser FC, Sproule JR, Halal F, Optiz JM. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet A. 1980;7:341–9.
Engels S, Kohlhase J, McGaughran J. ASALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet. 2000;37:458–60.
Morisada N, Sekine T, Ishimori S, Tsuda M, Adachi M, Nozu K, et al. 16q12 microdeletion syndrome in two Japanese boys. Pediatr Int. 2014;56:e75–8.
Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE, et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23:582–9.
Yoshikawa T, Kamei K, Nagata H, Saida K, Sato M, Ogura M, et al. Diversity of renal phenotypes in patients with WDR19 mutations: two case reports. Nephrology. 2017;22:566–71.
Ogawa A, Kitamura S, Nakayama K, Sugiyama H, Morisada N, Iijima K, et al. Right hypoplastic kidney. Kidney Int. 2012;82:1037.
Iwaki T, Wakabayashi T, Inoue A, Irie K, Fuke N, Kondo T, et al. A case of BOR syndrome complicated by cerebral cavernous malformation exhibiting novel mutation of the EYA1. Jpn. J. Pediatr. Nephrol. (in press).
Krug P, Morinière V, Marlin S, Koubi V, Gabriel HD, Colin E, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum Mutat. 2011;32:183–90.
Okada M, Fujimaru R, Morimoto N, Satomura K, Kaku Y, Tsuzuki K, et al. EYA1 and SIX1 gene mutations in Japanese patients with branchio-oto-renal (BOR) syndrome and related conditions. Pediatr Nephrol. 2006;21:475–81.
Wang SH, Wu CC, Lu YC, Lin YH, Su YN, Hwu WL, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in an East Asian cohort with branchio-oto-renal syndrome. Laryngoscope. 2012;122:1130–6.
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum Mol Genet. 1997;6:2247–55.
Morisada N, Rendtorff ND, Nozu K, Morishita T, Miyakawa T, Matsumoto T, et al. Branchio-oto-renal syndrome caused by partial EYA1 deletion due to LINE-1 insertion. Pediatr Nephrol. 2010;25:1343–8.
Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol. 2008;19:891–903.
Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85:1429–33.
Xu J, Wong EY, Cheng C, Li J, Sharkar MT, Xu CY, et al. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell. 2014;31:434–47.
Nishinakamura R, Matsumoto Y, Nakao K, Nakamura K, Sato A, Copeland NG, et al. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128:3105–15.
Kohlhase J. In: Townes-Brocks syndrome. Pagon RA, Adam MP, Ardinger HH, (eds). GeneReviews. Seattle: University of Washington; 2016.
Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80:800–4.
Nicolaou N, Pulit SL, Nijman IJ, Monroe GR, Feitz WF, Schreuder MF, et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 2016;89:476–86.
Acknowledgements
We thank all the participants and their families. We also thank Takashi Sekine (Toho University), Yoshitsugu Kaku, Pin Fee Chong (Fukuoka Children’s Hospital), Ayu Ogawa (Okayama University), Seiji Tanaka (Kurume University), Tsukasa Takemura (Kindai University), Kotaro Nomura (St Luke’s International Hospital), Midori Awazu (Keio University), Kazuaki Matsumoto (JA Toride Medical Center), Takahisa Yoshikawa (Kyoto University), Chikahiko Numakura (Yamagata University), Kei Nishiyama (Kyushu University), and Toshihiko Shirakawa (Nagasaki University) for their contributions. Furthermore, we are profoundly grateful to Tetsuko Yamanouchi and Yoshimi Nozu (Kobe University) for their technical assistance. We would like to thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
This work was supported by a Health Labor Sciences Research Grant for the Research on Measures for Intractable Diseases (H21-nanchi-ippan-103 to K.I., H24-nanchi-ippan-041 to K.I., and H29-nanchi-ippan-039 to N.M.) and Japan Society for the Promotion of Science KAKENHI grant number JP15K09261 (to N.M.). The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Unzaki, A., Morisada, N., Nozu, K. et al. Clinically diverse phenotypes and genotypes of patients with branchio-oto-renal syndrome. J Hum Genet 63, 647–656 (2018). https://doi.org/10.1038/s10038-018-0429-8
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s10038-018-0429-8
This article is cited by
-
Branchio-oto-renal syndrome in a young Han Chinese female: a case report and review of the literature
Journal of Medical Case Reports (2025)
-
Systematic genetic assessment of hearing loss using whole-genome sequencing identifies pathogenic variants
Experimental & Molecular Medicine (2025)
-
In vitro generation of a ureteral organoid from pluripotent stem cells
Nature Communications (2025)
-
Renal dysplasia development and chronic kidney disease
Pediatric Research (2025)
-
Clinical characteristics of patients with SALL1-related disorder
Pediatric Nephrology (2025)
