
ABSTRACT
Background
Congenital disorders of glycosylation (CDG) includes ALG8 deficiency, a protein N-glycosylation defect with a broad clinical spectrum. If most of the 15 previously reported patients present an early-onset multisystem severe disease and early death, three patients including the cas princeps, present long-term survival and less severe symptoms.
Methods
In order to further characterize ALG8-CDG, two new ALG8 patients are described and mRNA analyses of the ALG8-CDG cas princeps were effected.
Results
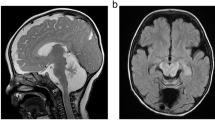
One new patient exhibited a hepato-intestinal and neurological phenotype with two novel variants (c.91A > C p.Thr31Pro; c.139dup p.Thr47Asnfs*12). The other new patient, homozygous for a known variant (c.845C > T p.Ala282Val), presented a neurological phenotype with epilepsy, intellectual disability and retinis pigmentosa. The cas princeps ALG8-CDG patient was reported to have two heterozygous frameshift variants predicted to be without activity. We now described a novel ALG8 transcript variant in this patient and the 3D model of the putative encoded protein reveals no major difference with that of the normal ALG8 protein.
Conclusion
The description of the two new ALG8 patients affirms that ALG8-CDG is a severe disease. In the cas princeps, as the originally described frameshift variants are degraded, the novel variant is promoted and could explain a milder phenotype.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Jaeken, J. & Matthijs, G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu. Rev. Genom. Hum. Genet. 8, 261–278 (2007).
Peanne, R. et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 30494–9 (2017).
Scott, K., Gadomski, T., Kozicz, T. & Morava, E. Congenital disorders of glycosylation: new defects and still counting. J. Inherit. Metab. Dis. 37, 609–617 (2014).
Ferreira, C. R. et al. Recognizable phenotypes in CDG. J. Inherit. Metab. Dis. 41, 541–553 (2018).
Schiff, M. et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J. Med. Genet. 54, 843–851 (2017).
Chantret, I. et al. A deficiency in dolichyl-P-glucose: Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new type of congenital disorders od glycosylation (CDG). J. Biol. Chem. 278, 9962–9971 (2003).
Charlwood, J. et al. A case of the carbohydrate-deficient glycoprotein syndrome type 1 (CDGS type 1) with normal phosphomannomutase activity. J. Inherit. Metab. Dis. 20, 817–826 (1997).
Eklund, E. A. et al. Congenital disorder of glycosylation (CDG)-Ih patient with a severe hepato-intestinal phenotype and evolving central nervous system pathology. J. Pediatr. 147, 847–850 (2005).
Schollen, E. et al. Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency). J. Med. Genet. 41, 550–556 (2004).
Vesela, K. et al. A new case of ALG8 deficiency (CDG Ih). J. Inherit. Metab. Dis 32, (Suppl 1), 259–264 (2009).
Stolting, T. et al. Novel ALG8 mutations expand the clinical spectrum of congenital disorder of glycosylation type Ih. Mol. Genet. Metab. 98, 305–309 (2009).
Bastaki, F. et al. Single-center experience of N-linked congenital disorders of glycosylation with a summary of molecularly characterized cases in Arabs. Ann. Hum. Genet. 82, 35–47 (2018).
Hock, M. et al. ALG8-CDG: novel patients and review of the literature. Orphanet J. Rare Dis. 10, 73 (2015).
Noensie, E. N. & Dietz, H. C. A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat. Biotechnol. 19, 434–439 (2001).
Ramensky, V., Bork, P. & Sunyaev, S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900 (2002).
Thomas, P. D. et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 (2003).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Tavtigian, S. V., Pierotti, M. A. & Borresen-Dale, A. L. International Agency for Research on Cancer Workshop on ‘Expression array analyses in breast cancer taxonomy’. Breast Cancer Res. 8, 303 (2006).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Lindahl, E., Azuara, C., Koehl, P. & Delarue, M. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 34, W52–W56 (2006).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Frischmeyer, P. A. & Dietz, H. C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 8, 1893–1900 (1999).
Hiller, M. et al. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat. Genet. 36, 1255–1257 (2004).
Hiller, M. & Platzer, M. Widespread and subtle: alternative splicing at short-distance tandem sites. Trends Genet 24, 246–255 (2008).
Dou, Y., Fox-Walsh, K. L., Baldi, P. F. & Hertel, K. J. Genomic splice-site analysis reveals frequent alternative splicing close to the dominant splice site. RNA 12, 2047–2056 (2006).
Ermakova, E. O., Nurtdinov, R. N. & Gelfand, M. S. Overlapping alternative donor splice sites in the human genome. J. Bioinform. Comput. Biol. 5, 991–1004 (2007).
Hamid, F. M. & Makeyev, E. V. Emerging functions of alternative splicing coupled with nonsense-mediated decay. Biochem. Soc. Trans. 42, 1168–1173 (2014).
Acknowledgments
This work was supported by a grant from ANR-15RAR3-0004-06 under the frame of E-RARE-3, the ERA-Net for Research on Rare Diseases.
Author information
Authors and Affiliations
Contributions
All authors have been involved in conception and design, or analysis and interpretation of data, and drafting the article or revising it critically for important intellectual content, and give their agreement to submission. S.V.-B., J.D., T.D., N.S., S.M., and I,C, are involved in analysis and interpretation of biochemistry and molecular data. M.S., E.S., A.D.H.O. d.B., P.d.L. collected clinical data. F.M. analyzed exome data for patient 2. A. C. performed 3D modeling data for normal and mutated protein of patient 1.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Vuillaumier-Barrot, S., Schiff, M., Mattioli, F. et al. Wide clinical spectrum in ALG8-CDG: clues from molecular findings suggest an explanation for a milder phenotype in the first-described patient. Pediatr Res 85, 384–389 (2019). https://doi.org/10.1038/s41390-018-0231-5
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41390-018-0231-5
