
Abstract
Background
As clinical exome sequencing (CES) becomes more common, understanding which patients are most likely to benefit and in what manner is critical for the general pediatrics community to appreciate.
Methods
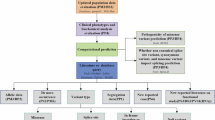
Five hundred and twenty-three patients referred to the Pediatric Genetics clinic at Michigan Medicine were systematically phenotyped by the presence or absence of abnormalities for 13 body/organ systems by a Clinical Genetics team. All patients then underwent CES.
Results
Overall, 30% of patients who underwent CES had an identified pathogenic mutation. The most common phenotypes were developmental delay (83%), neuromuscular system abnormalities (81%), and multiple congenital anomalies (42%). In all, 67% of patients had a variant of uncertain significance (VUS) or gene of uncertain significance (GUS); 23% had no variants reported. There was a significant difference in the average number of body systems affected among these groups (pathogenic 5.89, VUS 6.0, GUS 6.12, and no variant 4.6; P < 0.00001). Representative cases highlight four ways in which CES is changing clinical pediatric practice.
Conclusions
Patients with identified variants are enriched for multiple organ system involvement. Furthermore, our phenotyping provides broad insights into which patients are most likely to benefit from genetics referral and CES and how those results can help guide clinical practice more generally.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Gahl, W. A. et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet. Med. 14, 51–59 (2012).
Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Need, A. C. et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 49, 353–361 (2012).
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879 (2014).
Valencia, C. A. et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front. Pediatr. 3, 67 (2015).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511 (2013).
Clark, M. M. et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 3, 16 (2018).
Tan, T. Y. et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 171, 855–862 (2017).
Pena, L. D. M. et al. Looking beyond the exome: a phenotype-first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet. Med. 20, 464–469 (2018).
Sobreira, N. L. M. et al. Matchmaker exchange. Curr. Protoc. Hum. Genet. 95, 9.31. 31–39.31.15 (2017).
Boycott, K. M. et al. International cooperation to enable the diagnosis of all rare genetic diseases. Am. J. Hum. Genet. 100, 695–705 (2017).
Bick, D. et al. Successful application of whole genome sequencing in a medical genetics clinic. J. Pediatr. Genet. 6, 61–76 (2017).
Eldomery, M. K. et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 9, 26 (2017).
Cordoba, M. et al. Whole exome sequencing in neurogenetic odysseys: an effective, cost- and time-saving diagnostic approach. PLoS ONE 13, e0191228 (2018).
Posey, J. E. et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet. Med. 18, 678–685 (2016).
Trujillano, D. et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182 (2017).
Soden, S. E. et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med. 6, 265ra168 (2014).
van Maldergem, L., Wetzburger, C., Verloes, A., Fourneau, C. & Gillerot, Y. Mental retardation with blepharo-naso-facial abnormalities and hand malformations: a new syndrome? Clin. Genet. 41, 22–24 (1992).
Cappello, S. et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat. Genet. 45, 1300–1308 (2013).
Belligni, E. F., Palmer, R. W. & Hennekam, R. C. MECP2 duplication in a patient with congenital central hypoventilation. Am. J. Med. Genet. A. 152a, 1591–1593 (2010).
Acknowledgements
We thank the patients and their families, as well as the clerical staff in the Genetics Clinic, and the research coordinators of the University of Michigan Children’s Clinical Trial Support Unit: Chaandini Jayachandran, MS, CCRP; Alyssa Paul, MS, CCRC; Jonathon Wilbur, BS; Ciara Ivantics, MS; Amy Hurst, BS; Andy Brosius, BS. All phases of this study were supported by The University of Michigan, Department of Pediatrics and the Alfred A. Taubman Medical Research Institute. D.M.M. is also funded by the National Institutes of Health (NIH) DC R01-014456 and by the Donita B. Sullivan, MD, Research Professorship in Pediatrics. J.W.I. is supported by the Morton S. and Henrietta K. Sellner Professorship in Human Genetics.
Author information
Authors and Affiliations
Contributions
D.M.M., M.N.Z., J.W.I., and J.A.B. conceptualized and designed the study, coordinated and supervised data collection and data analysis, drafted the initial manuscript, and reviewed and revised the manuscript. A.A., M.C.H., C.E.K., and S.C.Q. directly evaluated patients, coordinated and supervised data collection, and critically reviewed the manuscript for intellectual content. N.W., R.F., M.G., J.E.J., K.N.L., T.B.M., J.D.O., B.C.O., L.S., L.T., S.B., and N.L.H. collected data and reviewed and revised the manuscript for intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ziats, M.N., Ahmad, A., Bernat, J.A. et al. Genotype–phenotype analysis of 523 patients by genetics evaluation and clinical exome sequencing. Pediatr Res 87, 735–739 (2020). https://doi.org/10.1038/s41390-019-0611-5
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41390-019-0611-5
This article is cited by
-
Contribution of families using the GenIDA database to the description of MED13L syndrome and literature review
Journal of Neurodevelopmental Disorders (2025)
-
Atypical Sotos syndrome caused by a novel splice site variant
Human Genome Variation (2022)
-
A Novel De Novo TUBB3 Variant Causing Developmental Delay, Epilepsy and Mild Ophthalmological Symptoms in a Chinese Child
Journal of Molecular Neuroscience (2022)
