
Abstract
Background
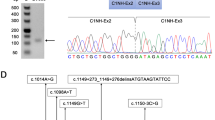
Loss-of-function variants in MID1 are the most common cause of Opitz G/BBB syndrome (OS). The interpretation of intronic variants affecting the splicing is a rising issue in OS.
Methods
Exon sequencing of a 2-year-old boy with OS showed that he was a carrier of the de novo c.1286–10G>T variant in MID1. In silico predictions and minigene assays explored the effect of the variant on splicing. The minigene approach was also applied to two previously identified MID1 c.864+1G>T and c.1285+1G>T variants.
Results
Minigene assay demonstrated that the c.1286–10G>T variant generated the inclusion of eight nucleotides that predicted generation of a frameshift. The c.864+1G>T and c.1285+1G>T variants resulted in an in-frame deletion predicted to generate a shorter MID1 protein. In hemizygous males, this allowed reclassification of all the identified variants from “of unknown significance” to “likely pathogenic.”
Conclusions
Minigene assay supports functional effects from MID1 intronic variants. This paves the way to the introduction of similar second-tier investigations in the molecular diagnostics workflow of OS.
Impact
-
Causative intronic variants in MID1 are rarely investigated in Opitz syndrome.
-
MID1 is not expressed in blood and mRNA studies are hardly accessible in routine diagnostics.
-
Minigene assay is an alternative for assessing the effect of intronic variants on splicing.
-
This is the first study characterizing the molecular consequences of three MID1 variants for diagnostic purposes and demonstrating the efficacy of minigene assays in supporting their clinical interpretation.
-
Review of the criteria according to the American College of Medical Genetics reassessed all variants as likely pathogenic.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Quaderi, N. A. et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 17, 285–291 (1997).
Kruszka, P. et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J. Med. Genet. 52, 104–110 (2015).
Robin, N. H. et al. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat. Genet. 11, 459–461 (1995).
Gaudenz, K. et al. Opitz G/BBB syndrome in Xp22: mutations in the MID1 gene cluster in the carboxy-terminal domain. Am. J. Hum. Genet. 63, 703–710 (1998).
Cox, T. C. et al. New mutations in MID1 provide support for loss of function as the cause of X-linked Opitz syndrome. Hum. Mol. Genet. 9, 2553–2562 (2000).
Pinson, L. et al. Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J. Med. Genet. 41, 381–386 (2004).
So, J. et al. Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations. Am. J. Med. Genet. A 132A, 1–7 (2005).
Mnayer, L., Khuri, S., Merheby, H. A., Meroni, G. & Elsas, L. J. A structure-function study of MID1 mutations associated with a mild Opitz phenotype. Mol. Genet. Metab. 87, 198–203 (2006).
Ferrentino, R., Bassi, M. T., Chitayat, D., Tabolacci, E. & Meroni, G. MID1 mutation screening in a large cohort of Opitz G/BBB syndrome patients: twenty-nine novel mutations identified. Hum. Mutat. 28, 206–207 (2007).
Reymond, A. et al. The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 (2001).
Trockenbacher, A. et al. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 29, 287–294 (2001).
Liu, E., Knutzen, C. A., Krauss, S., Schweiger, S. & Chiang, G. G. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc. Natl Acad. Sci. USA 108, 8680–8685 (2011).
Carracedo, A. & Pandolfi, P. P. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27, 5527–5541 (2008).
Short, K. M. & Cox, T. C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 281, 8970–8980 (2006).
Gholkar, A. A. et al. The X-linked-intellectual-disability-associated ubiquitin ligase Mid2 interacts with astrin and regulates astrin levels to promote cell division. Cell Rep. 14, 180–188 (2016).
Zanchetta, M. E., Napolitano, L. M. R., Maddalo, D. & Meroni, G. The E3 ubiquitin ligase MID1/TRIM18 promotes atypical ubiquitination of the BRCA2-associated factor 35, BRAF35. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1844–1854 (2017).
Zanchetta, M. E. & Meroni, G. Emerging roles of the TRIM E3 ubiquitin ligases MID1 and MID2 in cytokinesis. Front. Physiol. 10, 274 (2019).
Brnich, S. E. et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12, 3 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Tompson, S. W. & Young, T. L. Assaying the effects of splice site variants by exon trapping in a mammalian cell line. Bio Protoc. 7, e2281 (2017).
Abou, Tayoun et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Steffensen, A. Y. et al. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur. J. Hum. Genet. 22, 1362–1368 (2014).
Fraile-Bethencourt, E. et al. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 13, e1006691 (2017).
Giorgi, G. et al. Validation of CFTR intronic variants identified during cystic fibrosis population screening by a minigene splicing assay. Clin. Chem. Lab. Med. 53, 1719–1723 (2015).
Villate, O. et al. Functional analyses of a novel splice variant in the CHD7 gene, found by next generation sequencing, confirm its pathogenicity in a Spanish patient and diagnose him with CHARGE Syndrome. Front. Genet. 9, 7 (2018).
Morbidoni, V. et al. Hybrid minigene assay: an efficient tool to characterize mRNA splicing profiles of NF1 variants. Cancers 13, 999 (2021).
Hentze, M. W. & Kulozik, A. E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96, 307–310 (1999).
Linde, L. et al. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur. J. Hum. Genet. 15, 1156–1162 (2007).
Lejeune, F. Nonsense-mediated mRNA decay, a finely regulated mechanism. Biomedicines 10, 141 (2022).
Sarkar, H. et al. Nonsense-mediated mRNA decay efficiency varies in choroideremia providing a target to boost small molecule therapeutics. Hum. Mol. Genet. 28, 1865–1871 (2019).
Acknowledgements
The authors thank the families for their kind availability in sharing the findings within the scientific community. We acknowledge S.W. Tompson (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison) for providing pSPL3 vector.
Funding
This work was supported by the Ricerca Corrente 2018–2020 Program from the Italian Ministry of Health and Regione Puglia.
Author information
Authors and Affiliations
Contributions
L.M., G.M. and M.C. conceived and designed the work that led to the submission, acquired data, and played an important role in interpreting the results. F.R., M.M., G.N. and C.F. conducted functional studies. L.B. and S.M. analyzed the exome-sequencing data. L.M., G.M. and M.C. wrote the manuscript. All authors revised the manuscript. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
COMPETING interests
The authors declare no competing interests.
Ethics approval and consent to participate
Written informed consent was obtained from all individuals for publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Micale, L., Russo, F., Mascaro, M. et al. Opitz syndrome: improving clinical interpretation of intronic variants in MID1 gene. Pediatr Res 93, 1208–1215 (2023). https://doi.org/10.1038/s41390-022-02237-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41390-022-02237-y
