
Abstract
The discovery of active compounds for novel, underexplored targets is essential for advancing innovative therapeutics across a wide range of diseases. Recent advancements in artificial intelligence (AI) are revolutionizing active compound discovery by dramatically enhancing the efficiency, accuracy, and scalability previously challenged by traditional methods. This review provides a comprehensive overview of AI-driven methodologies for active compound discovery, with a particular focus on their application to novel targets. Initially, we explore how AI overcomes traditional bottlenecks in molecular design, enabling precise protein perception through high-accuracy protein structure prediction and enhanced docking precision. Building upon these target-focused capabilities, AI-driven approaches also advance ligand exploration, effectively bridging biological and chemical spaces through sophisticated data transfer techniques that maximize the utility of available activity data. By assessing overall cellular or organismal responses, AI plays a pivotal role in decoding complex biological systems, driving phenotypic drug discovery (PDD) through multi-modal data integration. Finally, we discuss how AI is addressing challenges associated with targeting previously undruggable proteins, exemplified by the development of protein degraders. By synthesizing these cutting-edge advancements, this review serves as a valuable resource for researchers seeking to leverage AI in the discovery of next-generation therapeutics.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
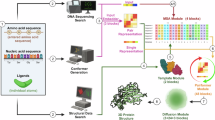
Data availability
This study does not involve the generation of new data. Therefore, data sharing is not applicable.
References
Zhong F, Xing J, Li X, Liu X, Fu Z, Xiong Z, et al. Artificial intelligence in drug design. Sci China Life Sci. 2018;61:1191–204.
Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022;12:3049–62.
Hwang TJ, Carpenter D, Lauffenburger JC, Wang B, Franklin JM, Kesselheim AS. Failure of investigational drugs in late-stage clinical development and publication of trial results. JAMA Intern Med. 2016;176:1826.
Xie X, Yu T, Li X, Zhang N, Foster LJ, Peng C, et al. Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials. Signal Transduct Target Ther. 2023;8:1–71.
Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21:181–200.
Sasso JM, Tenchov R, Wang D, Johnson LS, Wang X, Zhou QA. Molecular glues: the adhesive connecting targeted protein degradation to the clinic. Biochemistry. 2023;62:601–23.
Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7:93.
Chen J, Kriwacki RW. Intrinsically disordered proteins: structure, function and therapeutics. J Mol Biol. 2018;430:2275–7.
Bonomi M, Heller GT, Camilloni C, Vendruscolo M. Principles of protein structural ensemble determination. Curr Opin Struct Biol. 2017;42:106–16.
Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–606.
Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer. 2020;1:235–48.
Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:83.
Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, et al. TTD: therapeutic target database describing target druggability information. Nucleic Acids Res. 2024;52:D1465–77.
Brown DG, Wobst HJ. A decade of FDA-approved drugs (2010–2019): trends and future directions. J Med Chem. 2021;64:2312–38.
Sabe VT, Ntombela T, Jhamba LA, Maguire GEM, Govender T, Naicker T, et al. Current trends in computer-aided drug design and a highlight of drugs discovered via computational techniques: a review. Eur J Med Chem. 2021;224:113705.
Ren F, Ding X, Zheng M, Korzinkin M, Cai X, Zhu W, et al. AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor. Chem Sci. 2023;14:1443–52.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–6.
Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. 2023;379:1123–30.
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493–500.
Krishna R, Wang J, Ahern W, Sturmfels P, Venkatesh P, Kalvet I, et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science. 2024;384:eadl2528.
Bryant P, Kelkar A, Guljas A, Clementi C, Noé F. Structure prediction of protein-ligand complexes from sequence information with Umol. Nat Commun. 2024;15:4536.
Corso G, Stärk H, Jing B, Barzilay R, Jaakkola T. DiffDock: diffusion steps, twists, and turns for molecular docking. arXiv; 2022. Available from: https://arxiv.org/abs/2210.01776.
Masters MR, Mahmoud AH, Lill MA. Do deep learning models for co-folding learn the physics of protein-ligand interactions? Bioinformatics. 2024. Available from: http://biorxiv.org/lookup/doi/10.1101/2024.06.03.597219.
Brotzakis ZF, Zhang S, Murtada MH, Vendruscolo M. AlphaFold prediction of structural ensembles of disordered proteins. Nat Commun. 2025;16:1632.
Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock vina 1.2.0: new docking methods, expanded force field, and Python bindings. J Chem Inf Model. 2021;61:3891–8.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49.
Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, et al. DOCK 6: impact of new features and current docking performance. J Comput Chem. 2015;36:1132–56.
Wallach I, Dzamba M, Heifets A. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv; 2015. Available from: https://arxiv.org/abs/1510.02855.
Shen C, Zhang X, Deng Y, Gao J, Wang D, Xu L, et al. Boosting protein–ligand binding pose prediction and virtual screening based on residue–atom distance likelihood potential and graph transformer. J Med Chem. 2022;65:10691–706.
Cao D, Chen G, Jiang J, Yu J, Zhang R, Chen M, et al. Generic protein–ligand interaction scoring by integrating physical prior knowledge and data augmentation modelling. Nat Mach Intell. 2024;6:688–700.
Li Y, Li L, Wang S, Tang X. EQUIBIND: a geometric deep learning-based protein-ligand binding prediction method. Drug Discov Ther. 2023;17:363–4.
Lu W, Wu Q, Zhang J, Rao J, Li C, Zheng S. Tankbind: Trigonometry-aware neural networks for drug-protein binding structure prediction. Adv Neural Inf Process Syst. 2022;35:7236–49.
Cao D, Chen M, Zhang R, Wang Z, Huang M, Yu J, et al. SurfDock is a surface-informed diffusion generative model for reliable and accurate protein–ligand complex prediction. Nat Methods. 2025;22:310–22.
Prašnikar E, Ljubič M, Perdih A, Borišek J. Machine learning heralding a new development phase in molecular dynamics simulations. Artif Intell Rev. 2024;57:102.
Wang T, He X, Li M, Li Y, Bi R, Wang Y, et al. Ab initio characterization of protein molecular dynamics with AI2BMD. Nature. 2024;635:1019–27.
Ni Y, Feng S, Hong X, Sun Y, Ma WY, Ma ZM, et al. Pre-training with fractional denoising to enhance molecular property prediction. Nat Mach Intell. 2024;6:1169–78.
Guo AZ, Sevgen E, Sidky H, Whitmer JK, Hubbell JA, De Pablo JJ. Adaptive enhanced sampling by force-biasing using neural networks. J Chem Phys. 2018;148:134108.
Comer J, Gumbart JC, Hénin J, Lelièvre T, Pohorille A, Chipot C. The adaptive biasing force method: everything you always wanted to know but were afraid to ask. J Phys Chem B. 2015;119:1129–51.
Chen X, Wang K, Chen J, Wu C, Mao J, Song Y, et al. Integrative residue-intuitive machine learning and MD approach to unveil allosteric site and mechanism for β2AR. Nat Commun. 2024;15:8130.
Do HN, Wang J, Bhattarai A, Miao Y. GLOW: a workflow integrating gaussian-accelerated molecular dynamics and deep learning for free energy profiling. J Chem Theory Comput. 2022;18:1423–36.
Bhowmik D, Gao S, Young MT, Ramanathan A. Deep clustering of protein folding simulations. BMC Bioinforma. 2018;19:484.
Brandt S, Sittel F, Ernst M, Stock G. Machine learning of biomolecular reaction coordinates. J Phys Chem Lett. 2018;9:2144–50.
Lemke T, Peter C. EncoderMap: dimensionality reduction and generation of molecule conformations. J Chem Theory Comput. 2019;15:1209–15.
Lewis S, Hempel T, Jiménez Luna J, Gastegger M, Xie Y, Foong AYK, et al. Scalable emulation of protein equilibrium ensembles with generative deep learning. Mol Biol. 2024. Available from: http://biorxiv.org/lookup/doi/10.1101/2024.12.05.626885.
Karamzadeh R, Karimi-Jafari MH, Sharifi-Zarchi A, Chitsaz H, Salekdeh GH, Moosavi-Movahedi AA. Machine learning and network analysis of molecular dynamics trajectories reveal two chains of red/Ox-specific residue interactions in human protein disulfide isomerase. Sci Rep. 2017;7:3666.
Ward MD, Zimmerman MI, Meller A, Chung M, Swamidass SJ, Bowman GR. Deep learning the structural determinants of protein biochemical properties by comparing structural ensembles with DiffNets. Nat Commun. 2021;12:3023.
Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure–activity relationships. J Chem Inf Model. 2015;55:263–74.
Rose D, Wieder O, Seidel T, Langer T. PharmacoMatch: efficient 3D pharmacophore screening through neural subgraph matching. arXiv; 2024. Available from: http://arxiv.org/abs/2409.06316.
Suo Y, Qian X, Xiong Z, Liu X, Wang C, Mu B, et al. Enhancing the predictive power of machine learning models through a chemical space complementary DEL screening strategy. J Med Chem. 2024;67:18969–80.
Li X, Fourches D. Inductive transfer learning for molecular activity prediction: next-Gen QSAR models with MolPMoFiT. J Cheminformatics. 2020;12:27.
Upadhyay R, Phlypo R, Saini R, Liwicki M. Sharing to learn and learning to share; fitting together meta, multi-task, and transfer learning: a meta review. IEEE Access. 2024;12:148553–76.
Ross J, Belgodere B, Chenthamarakshan V, Padhi I, Mroueh Y, Das P. Large-scale chemical language representations capture molecular structure and properties. Nat Mach Intell. 2022;4:1256–64.
Cao Z, Sciabola S, Wang Y. Large-scale pretraining improves sample efficiency of active learning-based virtual screening. J Chem Inf Model. 2024;64:1882–91.
Chen S, Zhong F. GPCRSPACE: a new GPCR real expanded library based on large language models architecture and positive sample machine learning strategies. J Med Chem. 2024;67:16912–22.
Zhu J, Xia Y, Wu L, Xie S, Zhou W, Qin T, et al. Dual-view Molecular Pre-training. In: Proceedings of the 29th ACM SIGKDD Conference on Knowledge Discovery and Data Mining. New York, NY, USA: Association for Computing Machinery; 2023. p. 3615–27. (KDD ’23). Available from: https://doi.org/10.1145/3580305.3599317.
Pei Q, Wu L, Zhu J, Xia Y, Xie S, Qin T, et al. Breaking the barriers of data scarcity in drug–target affinity prediction. Brief Bioinform. 2023;24:bbad386.
Li Z, Li X, Liu X, Fu Z, Xiong Z, Wu X, et al. KinomeX: a web application for predicting kinome-wide polypharmacology effect of small molecules. Bioinformatics. 2019;35:5354–6.
Li Z, Qu N, Zhou J, Sun J, Ren Q, Meng J, et al. KinomeMETA: a web platform for kinome-wide polypharmacology profiling with meta-learning. Nucleic Acids Res. 2024;52:W489–97.
Lv Q, Chen G, Yang Z, Zhong W, Chen CYC. Meta learning with graph attention networks for low-data drug discovery. IEEE Trans Neural Netw Learn Syst. 2024;35:11218–30.
Wu Y, Xie L, Liu Y, Xie L. Semi-supervised meta-learning elucidates understudied molecular interactions. Commun Biol. 2024;7:1104.
Adams K, Abeywardane K, Fromer J, Coley CW. ShEPhERD: diffusing shape, electrostatics, and pharmacophores for bioisosteric drug design. arXiv; 2025. Available from: http://arxiv.org/abs/2411.04130.
Wang L, Wang S, Yang H, Li S, Wang X, Zhou Y, et al. Conformational space profiling enhances generic molecular representation for AI‐powered ligand‐based drug discovery. Adv Sci. 2024;11:2403998.
Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–35.
Madhukar NS, Khade PK, Huang L, Gayvert K, Galletti G, Stogniew M, et al. A Bayesian machine learning approach for drug target identification using diverse data types. Nat Commun. 2019;10:5221.
Zhao T, Hu Y, Valsdottir LR, Zang T, Peng J. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief Bioinform. 2021;22:2141–50.
Zhong F, Wu X, Yang R, Li X, Wang D, Fu Z, et al. Drug target inference by mining transcriptional data using a novel graph convolutional network framework. Protein Cell. 2022;13:281–301.
Chen H, King FJ, Zhou B, Wang Y, Canedy CJ, Hayashi J, et al. Drug target prediction through deep learning functional representation of gene signatures. Nat Commun. 2024;15:1853.
Himmelstein DS, Zietz M, Rubinetti V, Kloster K, Heil BJ, Alquaddoomi F, et al. Hetnet connectivity search provides rapid insights into how biomedical entities are related. GigaScience. 2023;12:giad047.
Zhang Y, Sui X, Pan F, Yu K, Li K, Tian S, et al. A comprehensive large-scale biomedical knowledge graph for AI-powered data-driven biomedical research. Nat Mach Intell. 2025;7:602–14.
Zheng S, Rao J, Song Y, Zhang J, Xiao X, Fang EF, et al. PharmKG: a dedicated knowledge graph benchmark for bomedical data mining. Brief Bioinform. 2021;22:bbaa344.
Chandak P, Huang K, Zitnik M. Building a knowledge graph to enable precision medicine. Sci Data. 2023;10:67.
Zeng X, Zhu S, Lu W, Liu Z, Huang J, Zhou Y, et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem Sci. 2020;11:1775–97.
Ni S, Kong X, Zhang Y, Chen Z, Wang Z, Fu Z, et al. Identifying compound-protein interactions with knowledge graph embedding of perturbation transcriptomics. Cell Genomics. 2024;4. Available from: https://www.cell.com/cell-genomics/abstract/S2666-979X(24)00266-0.
Song Q, Li M, Li Q, Lu X, Song K, Zhang Z, et al. DeepAlloDriver: a deep learning-based strategy to predict cancer driver mutations. Nucleic Acids Res. 2023;51:W129–33.
Kamimoto K, Stringa B, Hoffmann CM, Jindal K, Solnica-Krezel L, Morris SA. Dissecting cell identity via network inference and in silico gene perturbation. Nature. 2023;614:742–51.
Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6.
Zhu J, Wang J, Wang X, Gao M, Guo B, Gao M, et al. Prediction of drug efficacy from transcriptional profiles with deep learning. Nat Biotechnol. 2021;39:1444–52.
Lotfollahi M, Klimovskaia Susmelj A, De Donno C, Hetzel L, Ji Y, Ibarra IL, et al. Predicting cellular responses to complex perturbations in high‐throughput screens. Mol Syst Biol. 2023;19:e11517.
Roohani Y, Huang K, Leskovec J. Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nat Biotechnol. 2024;42:927–35.
Tong X, Qu N, Kong X, Ni S, Zhou J, Wang K, et al. Deep representation learning of chemical-induced transcriptional profile for phenotype-based drug discovery. Nat Commun. 2024;15:5378.
Yang F, Wang W, Wang F, Fang Y, Tang D, Huang J, et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat Mach Intell. 2022;4:852–66.
Theodoris CV, Xiao L, Chopra A, Chaffin MD, Al Sayed ZR, Hill MC, et al. Transfer learning enables predictions in network biology. Nature. 2023;618:616–24.
Hao M, Gong J, Zeng X, Liu C, Guo Y, Cheng X, et al. Large-scale foundation model on single-cell transcriptomics. Nat Methods. 2024;21:1481–91.
Cui H, Wang C, Maan H, Pang K, Luo F, Duan N, et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat Methods. 2024;21:1470–80.
Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc. 2016;11:1757–74.
Rohban MH, Fuller AM, Tan C, Goldstein JT, Syangtan D, Gutnick A, et al. Virtual screening for small-molecule pathway regulators by image-profile matching. Cell Syst. 2022;13:724–736.e9.
Simm J, Klambauer G, Arany A, Steijaert M, Wegner JK, Gustin E, et al. Repurposing high-throughput image assays enables biological activity prediction for drug discovery. Cell Chem Biol. 2018;25:611–618.e3.
Hofmarcher M, Rumetshofer E, Clevert DA, Hochreiter S, Klambauer G. Accurate prediction of biological assays with high-throughput microscopy images and convolutional networks. J Chem Inf Model. 2019;59:1163–71.
Bray MA, Gustafsdottir SM, Rohban MH, Singh S, Ljosa V, Sokolnicki KL, et al. A dataset of images and morphological profiles of 30,000 small-molecule treatments using the Cell Painting assay. GigaScience. 2017;6:giw014.
Zdrazil B, Felix E, Hunter F, Manners EJ, Blackshaw J, Corbett S, et al. The ChEMBL Ddatabase in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024;52:D1180–92.
Kusumoto D, Seki T, Sawada H, Kunitomi A, Katsuki T, Kimura M, et al. Anti-senescent drug screening by deep learning-based morphology senescence scoring. Nat Commun. 2021;12:257.
Janssens R, Zhang X, Kauffmann A, De Weck A, Durand EY. Fully unsupervised deep mode of action learning for phenotyping high-content cellular images. Cowen L, editor. Bioinformatics. 2021;37:4548–55.
Caie PD, Walls RE, Ingleston-Orme A, Daya S, Houslay T, Eagle R, et al. High-content phenotypic profiling of drug response signatures across distinct cancer cells. Mol Cancer Ther. 2010;9:1913–26.
Perakis A, Gorji A, Jain S, Chaitanya K, Rizza S, Konukoglu E. Contrastive learning of single-cell phenotypic representations for treatment classification. In: Lian C, Cao X, Rekik I, Xu X, Yan P, editors. Machine Learning in Medical Imaging. Cham: Springer International Publishing; 2021. p. 565–75.
Lu SZ, Lu Z, Hajiramezanali E, Biancalani T, Bengio Y, Scalia G, et al. Cell morphology-guided small molecule generation with GFlowNets. arXiv; 2024. Available from: https://arxiv.org/abs/2408.05196.
Marin Zapata PA, Méndez-Lucio O, Le T, Beese CJ, Wichard J, Rouquié D, et al. Cell morphology-guided de novo hit design by conditioning GANs on phenotypic image features. Digit Discov. 2023;2:91–102.
Lazo JS, Sharlow ER. Drugging undruggable molecular cancer targets. Annu Rev Pharm Toxicol. 2016;56:23–40.
Duran-Frigola M, Cigler M, Winter GE. Advancing targeted protein degradation via multiomics profiling and artificial intelligence. J Am Chem Soc. 2023;145:2711–32.
Troup RI, Fallan C, Baud MGJ. Current strategies for the design of PROTAC linkers: a critical review. Explor Target Antitumor Ther. 2020;1:273–312.
Abbas A, Ye F. Computational methods and key considerations for in silico design of proteolysis targeting chimera (PROTACs). Int J Biol Macromol. 2024;277:134293.
Igashov I, Stärk H, Vignac C, Schneuing A, Satorras VG, Frossard P, et al. Equivariant 3D-conditional diffusion model for molecular linker design. Nat Mach Intell. 2024;6:417–27.
Imrie F, Bradley AR, Van Der Schaar M, Deane CM. Deep generative models for 3D linker design. J Chem Inf Model. 2020;60:1983–95.
Tan Y, Dai L, Huang W, Guo Y, Zheng S, Lei J, et al. DRlinker: deep reinforcement learning for optimization in fragment linking design. J Chem Inf Model. 2022;62:5907–17.
García Jiménez D, Rossi Sebastiano M, Vallaro M, Mileo V, Pizzirani D, Moretti E, et al. Designing soluble PROTACs: strategies and preliminary guidelines. J Med Chem. 2022;65:12639–49.
Apprato G, D’Agostini G, Rossetti P, Ermondi G, Caron G. In silico tools to extract the drug design information content of degradation data: the case of PROTACs targeting the androgen receptor. Molecules. 2023;28:1206.
Prael FJ, Cox J, Sturm N, Kutchukian P, Forrester WC, Michaud G, et al. Machine learning proteochemometric models for Cereblon glue activity predictions. Artif Intell Life Sci. 2024;6:100100.
Su Z, Xiao D, Xie F, Liu L, Wang Y, Fan S, et al. Antibody–drug conjugates: recent advances in linker chemistry. Acta Pharm Sin B. 2021;11:3889–907.
Kong X, Huang W, Liu Y. Conditional antibody design as 3D equivariant graph translation. arXiv; 2022. Available from: https://arxiv.org/abs/2208.06073.
Gao K, Wu L, Zhu J, Peng T, Xia Y, He L, et al. Incorporating pre-training paradigm for antibody sequence-structure co-design. arXiv; 2022. Available from: https://arxiv.org/abs/2211.08406.
Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature. 2023;620:1089–100.
He H, He B, Guan L, Zhao Y, Jiang F, Chen G, et al. De novo generation of SARS-CoV-2 antibody CDRH3 with a pre-trained generative large language model. Nat Commun. 2024;15:6867.
Tingle BI, Tang KG, Castanon M, Gutierrez JJ, Khurelbaatar M, Dandarchuluun C, et al. ZINC-22─A free multi-billion-scale database of tangible compounds for ligand discovery. J Chem Inf Model. 2023;63:1166–76.
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2023 update. Nucleic Acids Res. 2023;51:D1373–80.
Burley SK, Bhikadiya C, Bi C, Bittrich S, Chao H, Chen L, et al. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023;51:D488–508.
Varadi M, Bertoni D, Magana P, Paramval U, Pidruchna I, Radhakrishnan M, et al. AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024;52:D368–75.
Liu Z, Su M, Han L, Liu J, Yang Q, Li Y, et al. Forging the basis for developing protein–ligand interaction scoring functions. Acc Chem Res. 2017;50:302–9.
Liu T, Hwang L, Burley SK, Nitsche CI, Southan C, Walters WP, et al. BindingDB in 2024: a FAIR knowledgebase of protein-small molecule binding data. Nucleic Acids Res. 2025;53:D1633–44.
Zhang C, Zhang X, Freddolino PL, Zhang Y. BioLiP2: an updated structure database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2024;52:D404–12.
Stein RM, Yang Y, Balius TE, O’Meara MJ, Lyu J, Young J, et al. Property-unmatched decoys in docking benchmarks. J Chem Inf Model. 2021;61:699–714.
Tran-Nguyen VK, Jacquemard C, Rognan D. LIT-PCBA: an unbiased data set for machine learning and virtual screening. J Chem Inf Model. 2020;60:4263–73.
Ljosa V, Sokolnicki KL, Carpenter AE. Annotated high-throughput microscopy image sets for validation. Nat Methods. 2012;9:637–637.
Sypetkowski M, Rezanejad M, Saberian S, Kraus O, Urbanik J, Taylor J, et al. RxRx1: a dataset for evaluating experimental batch correction methods. arXiv; 2023. Available from: https://arxiv.org/abs/2301.05768.
Chandrasekaran SN, Cimini BA, Goodale A, Miller L, Kost-Alimova M, Jamali N, et al. Three million images and morphological profiles of cells treated with matched chemical and genetic perturbations. Nat Methods. 2024;21:1114–21.
Zheng M, Okawa S, Bravo M, Chen F, Martínez-Chantar ML, del Sol A. ChemPert: mapping between chemical perturbation and transcriptional response for non-cancer cells. Nucleic Acids Res. 2023;51:D877–89.
Replogle JM, Saunders RA, Pogson AN, Hussmann JA, Lenail A, Guna A, et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell. 2022;185:2559–2575.e28.
Jayatunga MKP, Xie W, Ruder L, Schulze U, Meier C. AI in small-molecule drug discovery: a coming wave? Nat Rev Drug Discov. 2022;21:175–6.
Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, et al. ECOD: an evolutionary classification of protein domains. PLOS Comput Biol. 2014;10:e1003926.
Corso G, Deng A, Fry B, Polizzi N, Barzilay R, Jaakkola T. Deep Confident Steps to New Pockets: Strategies for Docking Generalization. arXiv; 2024. Available from: http://arxiv.org/abs/2402.18396
Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019;569:503–8.
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Acknowledgements
This work was supported by the following grants: the Strategic Priority Research Program of the Chinese Academy of sciences (XDB0830000), National Natural Science Foundation of China (82204278, T2225002 and 82273855), SIMM-SHUTCM Traditional Chinese Medicine Innovation Joint Research Program (E2G805H), Shanghai Municipal Science and Technology Major Project, National Key Research and Development Program of China (2023YFC2305904 and 2022YFC3400504), Key Technologies R&D Program of Guangdong Province (2023B1111030004), Shanghai Sailing Program (24YF2755600), and the China Postdoctoral Science Foundation (2024M763421).
Author information
Authors and Affiliations
Contributions
XYW: Writing—Original draft. YChen: Writing—Original draft. YFL: Writing—Original Draft. CYW: Writing—Original Draft. MYL: Literature Search and Screening. CXY: Writing—Original Draft. YYZ: Writing—Original Draft. MHQ: Writing—Original Draft. YFS: Writing—Original Draft. XCT: Conceptualization and Supervision. MYZ: Conceptualization and Supervision. XTL: Writing—Review and Editing, Conceptualization, and Supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, Xy., Chen, Y., Li, Yf. et al. Advancing active compound discovery for novel drug targets: insights from AI-driven approaches. Acta Pharmacol Sin (2025). https://doi.org/10.1038/s41401-025-01591-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41401-025-01591-x
