
Abstract
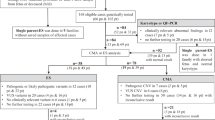
We performed whole exome or genome sequencing in eight multiply affected families with ostensibly isolated congenital anosmia. Hypothesis-free analyses based on the assumption of fully penetrant recessive/dominant/X-linked models obtained no strong single candidate variant in any of these families. In total, these eight families showed 548 rare segregating variants that were predicted to be damaging, in 510 genes. Three Kallmann syndrome genes (FGFR1, SEMA3A, and CHD7) were identified. We performed permutation-based analysis to test for overall enrichment of these 510 genes carrying these 548 variants with genes mutated in Kallmann syndrome and with a control set of genes mutated in hypogonadotrophic hypogonadism without anosmia. The variants were found to be enriched for Kallmann syndrome genes (3 observed vs. 0.398 expected, p = 0.007), but not for the second set of genes. Among these three variants, two have been already reported in genes related to syndromic anosmia (FGFR1 (p.(R250W)), CHD7 (p.(L2806V))) and one was novel (SEMA3A (p.(T717I))). To replicate these findings, we performed targeted sequencing of 16 genes involved in Kallmann syndrome and hypogonadotrophic hypogonadism in 29 additional families, mostly singletons. This yielded an additional 6 variants in 5 Kallmann syndrome genes (PROKR2, SEMA3A, CHD7, PROK2, ANOS1), two of them already reported to cause Kallmann syndrome. In all, our study suggests involvement of 6 syndromic Kallmann genes in isolated anosmia. Further, we report a yet unreported appearance of di-genic inheritance in a family with congenital isolated anosmia. These results are consistent with a complex molecular basis of congenital anosmia.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Karstensen HG, Tommerup N. Isolated and syndromic forms of congenital anosmia. Clin Genet. 2012;81:210–5.
Feldmesser E, Bercovich D, Avidan N, et al. Mutations in olfactory signal transduction genes are not a major cause of human congenital general anosmia. Chem Sense. 2007;32:21–30.
Dode C, Teixeira L, Levilliers J, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175.
Weiss J, Pyrski M, Jacobi E, et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature. 2011;472:186–90.
Kulaga HM, Leitch CC, Eichers ER, et al. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet. 2004;36:994–98.
McEwen DP, Koenekoop RK, Khanna H, et al. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc Natl Acad Sci U S A. 2007;104:15917–922.
Skjeldal OH, Stokke O, Refsum S, Norseth J, Petit H. Clinical and biochemical heterogeneity in conditions with phytanic acid accumulation. J Neurol Sci. 1987;77:87–96.
Karstensen HG, Mang Y, Fark T, Hummel T, Tommerup N. The first mutation in CNGA2 in two brothers with anosmia. Clin Genet. 2015;88:293–96.
Ghadami M, Morovvati S, Majidzadeh AK, et al. Isolated congenital anosmia locus maps to 18p11.23-q12.2. J Med Genet. 2004;41:299–303.
Moya-Plana A, Villanueva C, Laccourreye O, Bonfils P, de Roux N. PROKR2 and PROK2 mutations cause isolated congenital anosmia without gonadotropic deficiency. Eur J Endocrinol. 2013;168:31–37.
Alkelai A, Olender T, Haffner-Krausz R et al. A role for TENM1 mutations in congenital general anosmia. Clin Genet. 2016;90:211–19.
Mitchell AL, Dwyer A, Pitteloud N, Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trend Endocrinol Metab. 2011;22:249–58.
Hipkin LJ, Casson IF, Davis JC. Identical twins discordant for Kallmann’s syndrome. J Med Genet. 1990;27:198–99.
Pitteloud N, Quinton R, Pearce S, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 2007;117:457–63.
Pitteloud N, Meysing A, Quinton R, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006;254-255:60–69.
Kim SH: Congenital hypogonadotropic hypogonadism and kallmann syndrome: past, present, and future. Endocrinol Metab 2015; 30 : 456-66.
Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11:547–64.
Quaynor SD, Kim HG, Cappello EM, et al. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril. 2011;96:1424–30 e1426.
Dode C, Hardelin JP. Kallmann syndrome. Eur J Hum Genet. 2009;17:139–46.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–760.
Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–79.
McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acid Res. 2010;38:e164.
Liu X, Jian X, Boerwinkle E. dbNSFPv2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–2402.
Zhu M, Need AC, Han Y, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91:408–21.
Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–32.
Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73.
Keydar I, Ben-Asher E, Feldmesser E, et al. General olfactory sensitivity database (GOSdb): candidate genes and their genomic variations. Hum Mutat. 2013;34:32–41.
Stelzer G, Plaschkes I, Oz-Levi D et al. VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics 2016; In Press.
Goes FS, Pirooznia M, Parla JS, et al. Exome sequencing of familial bipolar disorder. JAMA Psychiatr. 2016;73:590–97.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709.
Rombaux P, Grandin C, Duprez T. How to measure olfactory bulb volume and olfactory sulcus depth? B-ENT. 2009;5:53–60.
Duprez TP, Rombaux P. Imaging the olfactory tract (cranial nerve #1). Eur J Radiol. 2010;74:288–98.
Marcos S, Sarfati J, Leroy C, et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab. 2014;99:E2138–43.
Hanchate NK, Giacobini P, Lhuillier P, et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 2012;8:e1002896.
Trarbach EB, Costa EM, Versiani B, et al. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab. 2006;91:4006–12.
Gu WJ, Zhang Q, Wang YQ, et al. Mutation analyses in pedigrees and sporadic cases of ethnic Han Chinese Kallmann syndrome patients. Exp Biol Med. 2015;240:1480–89.
Dode C, Fouveaut C, Mortier G, et al. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Human Mutat. 2007;28:97–98.
Vuorela P, Ala-Mello S, Saloranta C, et al. Molecular analysis of the CHD7 gene in CHARGE syndrome: identification of 22 novel mutations and evidence for a low contribution of large CHD7 deletions. Genet Med. 2007;9:690–94.
Bartels CF, Scacheri C, White L, Scacheri PC, Bale S. Mutations in the CHD7 gene: the experience of a commercial laboratory. Genet Test Mol Biomark. 2010;14:881–91.
Dode C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–65.
Young J, Metay C, Bouligand J, et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum Reprod. 2012;27:1460–65.
Waldstreicher J, Seminara SB, Jameson JL, et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81:4388–95.
Layman LC. Clinical genetic testing for Kallmann syndrome. J Clin Endocrinol Metab. 2013;98:1860–62.
Hardelin JP, Dode C. The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sex Dev. 2008;2:181–93.
Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71.
Belluscio L, Gold GH, Nemes A, Axel R. Mice deficient in G(olf) are anosmic. Neuron. 1998;20:69–81.
Wong ST, Trinh K, Hacker B, et al. Disruption of the type III adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron. 2000;27:487–97.
Brunet LJ, Gold GH, Ngai J. General anosmia caused by a targeted disruption of the mouse olfactory cyclic nucleotide-gated cation channel. Neuron. 1996;17:681–93.
Acknowledgements
This research was supported by Nella and Leon Benoziyo Center for Neurological Diseases, Weizmann Institute of Science; The Crown Human Genome Center, Weizmann Institute of Science; and by The Legacy Heritage Biomedical Science Partnership Program of the Israel Science Foundation, 586/11.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Alkelai, A., Olender, T., Dode, C. et al. Next-generation sequencing of patients with congenital anosmia. Eur J Hum Genet 25, 1377–1387 (2017). https://doi.org/10.1038/s41431-017-0014-1
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41431-017-0014-1
This article is cited by
-
Molecular genetic diagnostics of hypogonadotropic hypogonadism: from panel design towards result interpretation in clinical practice
Human Genetics (2021)
-
Genome analysis and knowledge-driven variant interpretation with TGex
BMC Medical Genomics (2019)
-
A novel FGF8 mutation in a female patient with isolated congenital anosmia
Hormones (2019)
