
Abstract
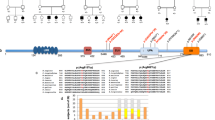
Intellectual disability (ID), megalencephaly, frontal predominant pachygyria, and seizures, previously called “thin” lissencephaly, are reported to be caused by recessive variants in CRADD. Among five families of different ethnicities identified, one homozygous missense variant, c.509G>A p.(Arg170His), was of Finnish ancestry. Here we report on the phenotypic variability associated for this potential CRADD founder variant in 22 Finnish individuals. Exome sequencing was used to identify candidate genes in Finnish patients presenting with ID. Targeted Sanger sequencing and restriction enzyme analysis were applied to screen for the c.509G>A CRADD variant in cohorts from Finland. Detailed phenotyping and genealogical studies were performed. Twenty two patients were identified with the c.509G>A p.(Arg170His) homozygous variant in CRADD. The majority of the ancestors originated from Northeastern Finland indicating a founder effect. The hallmark of the disease is frontotemporal predominant pachygyria with mild cortical thickening. All patients show ID of variable severity. Aggressive behavior was found in nearly half of the patients, EEG abnormalities in five patients and megalencephaly in three patients. This study provides detailed data about the phenotypic spectrum of patients with lissencephaly due to a CRADD variant that affects function. High inter- and intrafamilial phenotypic heterogeneity was identified in patients with pachygyria caused by the homozygous CRADD founder variant. The phenotype variability suggests that additional genetic and/or environmental factors play a role in the clinical presentation. Since frontotemporal pachygyria is the hallmark of the disease, brain imaging studies are essential to support the molecular diagnosis for individuals with ID and a CRADD variant.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Change history
10 September 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104.
Willemsen MH, Kleefstra T. Making headway with genetic diagnostics of intellectual disabilities. Clin Genet. 2014;85:101–10.
Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. 2017;22:1604–14.
Institute of Medicine (U.S.). Committee to evaluate the supplemental security income disability program for children with mental disorders. In: Boat TF, Wu JT, editors. National Academies of Sciences Engineering and Medicine: mental disorders and disabilities among low-income children. Washington, D.C.: National Academies Press, 2015. .
Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genom Hum Genet. 2010;11:161–87.
Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–69.
Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014;13:710–26.
Guerrini R, Marini C. Genetic malformations of cortical development. Exp Brain Res. 2006;173:322–33.
Di Donato N, Chiari S, Mirzaa GM, Aldinger K, Parrini E, Olds C, et al. Lissencephaly: expanded imaging and clinical classification. Am J Med Genet A. 2017;173:1473–88.
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–21.
Parrini E, Conti V, Dobyns WB, Guerrini R. Genetic basis of brain malformations. Mol Syndromol. 2016;7:220–33.
Fry AE, Cushion TD, Pilz DT. The genetics of lissencephaly. Am J Med Genet C Semin Med Genet. 2014;166C:198–210.
Di Donato N, Jean YY, Maga AM, Krewson BD, Shupp AB, Avrutsky MI, et al. Mutations in CRADD result in reduced caspase-2-mediated neuronal apoptosis and cause megalencephaly with a rare lissencephaly variant. Am J Hum Genet. 2016;99:1117–29.
Harel T, Hacohen N, Shaag A, Gomori M, Singer A, Elpeleg O, et al. Homozygous null variant in CRADD, encoding an adaptor protein that mediates apoptosis, is associated with lissencephaly. Am J Med Genet A. 2017;173:2539–44.
Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28936.
Kurki MI, Saarentaus E, Pietilainen O, Gormley P, Lal D, Kerminen S et al. Contribution of rare and common variants to intellectual disability in a high-risk population sub-isolate of Northern Finland. bioRxiv. 2019;10:410.
Avela K, Toiviainen-Salo S, Karttunen-Lewandowski P, Kauria L, Valanne L, Salonen-Kajander R. Frontotemporal pachygyria-two new patients. Eur J Med Genet. 2012;55:753–7.
Sorva R, Tolppanen EM, Lankinen S, Perheentupa J. [Evaluation of childhood growth]. Duodecim. 1985;101:465–76.
Karvonen M, Hannila ML, Saari A, Dunkel L. New Finnish reference for head circumference from birth to 7 years. Ann Med. 2012;44:369–74.
Saari A, Sankilampi U, Hannila ML, Kiviniemi V, Kesseli K, Dunkel L. New Finnish growth references for children and adolescents aged 0 to 20 years: Length/height-for-age, weight-for-length/height, and body mass index-for-age. Ann Med. 2011;43:235–48.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Sequencing Initiative Suomi project (SISu), Institute for Molecular Medicine Finland (FIMM), University of Helsinki, Finland. http://sisuproject.fi. [SISu v4.1, 03/2018].
Martin AR, Karczewski KJ, Kerminen S, Kurki MI, Sarin AP, Artomov M, et al. Haplotype sharing provides insights into fine-scale population history and disease in Finland. Am J Hum Genet. 2018;102:760–75.
Varilo T, Nikali K, Suomalainen A, Lonnqvist T, Peltonen L. Tracing an ancestral mutation: genealogical and haplotype analysis of the infantile onset spinocerebellar ataxia locus. Genome Res. 1996;6:870–5.
Duan H, Dixit VM. RAIDD is a new ‘death’ adaptor molecule. Nature. 1997;385:86–89.
Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet. 2000;26:122–7.
Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R, et al. Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc Natl Acad Sci USA. 2005;102:14314–20.
Ribe EM, Jean YY, Goldstein RL, Manzl C, Stefanis L, Villunger A, et al. Neuronal caspase 2 activity and function requires RAIDD, but not PIDD. Biochem J. 2012;444:591–9.
Shalini S, Dorstyn L, Wilson C, Puccini J, Ho L, Kumar S. Impaired antioxidant defence and accumulation of oxidative stress in caspase-2-deficient mice. Cell Death Differ. 2012;19:1370–80.
Zhang Y, Padalecki SS, Chaudhuri AR, De Waal E, Goins BA, Grubbs B, et al. Caspase-2 deficiency enhances aging-related traits in mice. Mech Ageing Dev. 2007;128:213–21.
Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. 1999;23:233–6.
Aula P, Autio S, Raivio KO, Rapola J, Thoden CJ, Koskela SL, et al. “Salla disease”: a new lysosomal storage disorder. Arch Neurol. 1979;36:88–94.
Renlund M, Aula P, Raivio KO, Autio S, Sainio K, Rapola J, et al. Salla disease: a new lysosomal storage disorder with disturbed sialic acid metabolism. Neurology. 1983;33:57–66.
Kure S, Takayanagi M, Narisawa K, Tada K, Leisti J. Identification of a common mutation in Finnish patients with nonketotic hyperglycinemia. J Clin Invest. 1992;90:160–4.
Peltonen L, Palotie A, Lange K. Use of population isolates for mapping complex traits. Nat Rev Genet. 2000;1:182–90.
Norio R. Finnish disease heritage I: characteristics, causes, background. Hum Genet. 2003;112:441–56.
Polvi A, Linturi H, Varilo T, Anttonen AK, Byrne M, Fokkema IF, et al. The Finnish disease heritage database (FinDis) update-a database for the genes mutated in the Finnish disease heritage brought to the next-generation sequencing era. Hum Mutat. 2013;34:1458–66.
Romero DM, Bahi-Buisson N, Francis F. Genetics and mechanisms leading to human cortical malformations. Semin. Cell Dev. Biol. 2017;76:33–75.
Acknowledgements
We are grateful to the families for participating in the study. We thank Shaffaq Raza, Minna Varhala, and Eija Hämäläinen for excellent technical help. We thank all the clinicians who have recruited patients to this study. This work was supported by the European Union’s Seventh Framework Program (Gencodys; grant 241995 to HvB). DLP is recipient of a CAPES Fellowship (99999.013311/2013–01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Polla, D.L., Rahikkala, E., Bode, M.K. et al. Phenotypic spectrum associated with a CRADD founder variant underlying frontotemporal predominant pachygyria in the Finnish population. Eur J Hum Genet 27, 1235–1243 (2019). https://doi.org/10.1038/s41431-019-0383-8
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41431-019-0383-8
This article is cited by
-
Bi-allelic truncating variants in CASP2 underlie a neurodevelopmental disorder with lissencephaly
European Journal of Human Genetics (2024)
-
Proteogenomic analysis of human cerebrospinal fluid identifies neurologically relevant regulation and implicates causal proteins for Alzheimer’s disease
Nature Genetics (2024)
-
Congenital hydrocephalus: new Mendelian mutations and evidence for oligogenic inheritance
Human Genomics (2023)
-
Pathogenic variants in PIDD1 lead to an autosomal recessive neurodevelopmental disorder with pachygyria and psychiatric features
European Journal of Human Genetics (2021)
-
Biallelic mutations in the death domain of PIDD1 impair caspase-2 activation and are associated with intellectual disability
Translational Psychiatry (2021)
