
Abstract
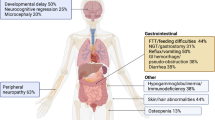
Clinical, pathological, and genetic findings of a primary hereditary ataxia found in a Malinois dog family are described and compared with its human counterpart. Based on the family history and the phenotype/genotype relationships already described in humans and dogs, a causal variant was expected to be found in KCNJ10. Rather surprisingly, whole-exome sequencing identified the SLC12A6 NC_006612.3(XM_014109414.2): c.178_181delinsCATCTCACTCAT (p.(Met60Hisfs*14)) truncating variant. This loss-of-function variant perfectly segregated within the affected Malinois family in an autosomal recessive way and was not found in 562 additional reference dogs from 18 different breeds, including Malinois. In humans, SLC12A6 variants cause “agenesis of the corpus callosum with peripheral neuropathy” (ACCPN, alias Andermann syndrome), owing to a dysfunction of this K+–Cl− cotransporter. However, depending on the variant (including truncating variants), different clinical features are observed within ACCPN. The variant in dogs encodes the shortest isoform described so far and its resultant phenotype is quite different from humans, as no signs of peripheral neuropathy, agenesis of the corpus callosum nor obvious mental retardation have been observed in dogs. On the other hand, progressive spinocerebellar ataxia, which is the most important feature of the canine phenotype, hindlimb paresis, and myokymia-like muscle contractions have not been described in humans with ACCPN so far. As this is the first report of a naturally occurring disease-causing SLC12A6 variant in a non-human species, the canine model will be highly valuable to better understand the complex molecular pathophysiology of SLC12A6-related neurological disorders and to evaluate novel treatment strategies.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Hytönen MK, Lohi H. Canine models of human rare disorders. Rare Dis. 2016;4:e1241362.3.
Wallace SE, Bird TD. Molecular genetic testing for hereditary ataxia: What every neurologist should know. Neurol Clin Pract. 2018;8:27–32.
Sandford E, Burmeister M. Genes and genetic testing in hereditary ataxias. Genes. 2014;5:586–603.
Gilliam D, O’Brien DP, Coates JR, Johnson GS, Johnson GC, Mhlanga-Mutangadura T, et al. A homozygous KCNJ10 mutation in Jack Russell Terriers and related breeds with spinocerebellar ataxia with myokymia, seizures, or both. J Vet Intern Med. 2014;28:871–7.
Van Poucke M, Stee K, Bhatti SF, Vanhaesebrouck A, Bosseler L, Peelman LJ, et al. The novel homozygous KCNJ10 c.986T>C (p.(Leu329Pro)) variant is pathogenic for the SeSAME/EAST homologue in Malinois dogs. Eur J Hum Genet. 2017;25:222–6.
Broeckx B, Hitte C, Coopman F, Verhoeven GE, De Keulenaer S, De Meester E, et al. Improved canine exome designs, featuring ncRNAs and increased coverage of protein coding genes. Sci Rep. 2015;5:12810.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122.
Broeckx B, Peelman L, Saunders J, Deforce D, Clement L. Using variant databases for variant prioritization and to detect erroneous genotype-phenotype associations. BMC Bioinformatics. 2017;18:535.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Van Poucke M, Martlé V, Van Brantegem L, Ducatelle R, Van Ham L, Bhatti S, et al. A canine orthologue of the human GFAP c.716G4A (p.Arg239His) variant causes Alexander disease in a Labrador retriever. Eur J Hum Genet. 2016;24:852–6.
Gast AC, Metzger J, Tipold A, Distl O. Genome-wide association study for hereditary ataxia in the Parson Russell Terrier and DNA-testing for ataxia-associated mutations in the Parson and Jack Russell Terrier. BMC Vet Res. 2016;12:225.
Garneau AP, Marcoux AA, Frenette-Cotton R, Mac-Way F, Lavoie JL, Isenring P. Molecular insights into the normal operation, regulation, and multisystemic roles of K + -Cl- cotransporter 3 (KCC3). Am J Physiol Cell Physiol. 2017;313:C516–C532.
Hiki K, D’Andrea RJ, Furze J, Crawford J, Woollatt E, Sutherland GR, et al. Cloning, characterization, and chromosomal location of a novel human K+-Cl- cotransporter. J Biol Chem. 1999;274:10661–7.
Mount DB, Mercado A, Song L, Xu J, George AL, Delpire E, et al. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem. 1999;274:16355–62.
Flores B, Schornack CC, Delpire E. A role for KCC3 in maintaining cell volume of peripheral nerve fibers. Neurochem Int. 2019;123:114–24.
Kahle KT, Khanna AR, Alper SL, Adragna NC, Lauf PK, Sun D, et al. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol Med. 2015;21:513–23.
Bowerman M, Salsac C, Bernard V, Soulard C, Dionne A, Coque E, et al. KCC3 loss-of-function contributes to Andermann syndrome by inducing activity-dependent neuromuscular junction defects. Neurobiol Dis. 2017;106:35–48.
Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384–92.
Larbrisseau A, Vanasse M, Brochu P, Jasmin G. The Andermann syndrome: agenesis of the corpus callosum associated with mental retardation and progressive sensorimotor neuronopathy. Can J Neurol Sci. 1984;11:257–61.
Hauser BE, Bittner R, Liegl C, Bernert G, Zeitlhofer J. Occurrence of Andermann syndrome out of French Canada – agenesis of the corpus callosum with neuronopathy. Neuropediatrics. 1993;24:107–10.
Filteau MJ, Pourcher E, Bouchard RH, Baruch P, Mathieu J, Bédard F, et al. Corpus callosum agenesis and psychosis in andermann syndrome. Arch Neurol. 1991;48:1275–80.
Deleu D, Bamanikar SA, Muirhead D, Louon A. Familial progressive sensorimotor neuropathy with agenesis of the corpus callosum (Andermann Syndrome): a clinical, neuroradiological and histopathological study. Eur Neurol. 1997;37:104–9.
Auer R, Laganière J, Robitaille Y, Richardson J, Dion PA, Rouleau GA, et al. KCC3 axonopathy: neuropathological features in the central and peripheral nervous system. Mod Pathol. 2016;29:962–76.
Salin-Cantegrel A, Shekarabi M, Holbert S, Dion P, Rochefort D, Laganière J, et al. HMSN/ACC truncation mutations disrupt brain-type creatine kinase-dependant activation of K+/Cl- co-transporter 3. Hum Mol Genet. 2008;17:2703–11.
Salin-Cantegrel A, Rivière JB, Shekarabi M, Rasheed S, Dacal S, Laganière J, et al. Transit defect of potassium-chloride Co-transporter 3 is a major pathogenic mechanism in hereditary motor and sensory neuropathy with agenesis of the corpus callosum. J Biol Chem. 2011;286:28456–65.
Uyanik G, Elcioglu N, Penzien J, Gross C, Yilmaz Y, Olmez A, et al. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology. 2006;66:1044–8.
Kahle KT, Flores B, Bharucha-Goebel D, Zhang J, Donkervoort S, Hegde M, et al. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci Signal. 2016;9:ra77.
Salin-Cantegrel A, Shekarabi M, Rasheed S, Charron FM, Laganière J, Gaudet R, et al. Potassium-chloride cotransporter 3 interacts with Vav2 to synchronize the cell volume decrease response with cell protrusion dynamics. PLoS ONE. 2013;8:e65294.
Ding J, Delpire E. Deletion of KCC3 in parvalbumin neurons leads to locomotor deficit in a conditional mouse model of peripheral neuropathy associated with agenesis of the corpus callosum. Behav Brain Res. 2014;274:128–36.
Bhatti SF, Vanhaesebrouck A, Van Soens I, Martlé VA, Polis IE, Rusbridge C, et al. Myokymia and neuromyotonia in 37 Jack Russell terriers. Vet J. 2011;189:284–8.
Rudnik-Schöneborn S, Hehr U, von Kalle T, Bornemann A, Winkler J, Zerres K. Andermann Syndrome can be a phenocopy of hereditary motor and sensory neuropathy – report of a discordant sibship with a compound heterozygous mutation of the KCC3 gene. Neuropediatrics. 2009;40:129–33.
Lourenço CM, Dupré N, Rivière JB, Rouleau GA, Marques VD, Genari AB, et al. Expanding the differential diagnosis of inherited neuropathies with non-uniform conduction: Andermann syndrome. J Peripher Nerv Syst. 2012;17:123–7.
Sinnwell JP, Therneau TM, Schaid DJ. The kinship2 R package for pedigree data. Hum Hered. 2014;78:91–93.
Löytynoja A, Goldman N. Short template switch events explain mutation clusters in the human genome. Genome Res. 2017;27:1039–49.
Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30:884–6.
Acknowledgements
We thank Sylvie Decraene, Sarah De Keulenaer, Ellen De Meester, Linda Impe, Caroline Rogiers, Dominique Vander Donckt, and Ruben Van Gansbeke for excellent technical assistance, Sameh A. Youssef and Martí Pumarola for their second opinion about the histological slides, as well as the breeders, owners, and veterinarians who collaborated to this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Van Poucke, M., Stee, K., Sonck, L. et al. Truncating SLC12A6 variants cause different clinical phenotypes in humans and dogs. Eur J Hum Genet 27, 1561–1568 (2019). https://doi.org/10.1038/s41431-019-0432-3
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41431-019-0432-3
