
Abstract
Myotonic dystrophy type 1 (DM1) is the most prevalent muscular dystrophy in adulthood and is one of the most clinically diverse monogenic diseases. Although it is classified as a neuromuscular disease, DM1 is a multisystem disorder that affects nearly all organ systems, particularly skeletal and smooth muscles, the central nervous system and the heart. Its phenotypic variability extends beyond a continuum of severity, encompassing differences in age of onset and organ involvement. DM1 is caused by a trinucleotide (CTG) repeat expansion within the 3′ untranslated region of the DMPK gene, leading to a toxic RNA gain-of-function mechanism that disrupts RNA splicing, causing widespread cellular dysfunction. Despite progress in understanding DM1 pathogenesis, gaps remain in elucidating genotype–phenotype correlations, genetic modifiers and mechanisms that influence disease progression. Breakthroughs in the past five to ten years have uncovered important insights into the molecular underpinnings of DM1 and accelerated therapeutic innovation. Targeted interventions such as small molecules, antisense oligonucleotides and gene-editing technologies are progressing into clinical trials. Additionally, emerging research on somatic instability, epigenetic modifications and novel biomarkers suggests approaches for precision medicine. This Review synthesizes recent clinical and molecular discoveries, highlighting implications for therapy development. By integrating clinical heterogeneity with mechanistic insights, we provide a framework for future translational research and therapeutic innovation in this life-limiting disease.
Key points
-
Myotonic dystrophy type 1 (DM1) is a severe, inherited multisystem disorder with marked variability in age at onset, organ involvement and severity, often showing genetic anticipation across generations.
-
A CTG repeat expansion in the dystrophia myotonica protein kinase (DMPK) gene produces toxic RNA that sequesters muscleblind-like (MBNL) proteins and increases activity of the RNA-binding protein CELF1, leading to widespread splicing defects.
-
Advances in understanding DM1 disease mechanisms increasingly link molecular pathology to clinical symptoms, though full genotype–phenotype correlations remain incomplete.
-
Therapeutic strategies that target the toxic RNA, restore the function of MBNL proteins or remove the CTG repeat expansion are advancing rapidly.
-
Clinical trial readiness efforts focus on improving outcome measures, identifying responsive subgroups and addressing disease heterogeneity to accelerate therapeutic development.
-
Clinical trials of antisense oligonucleotides, small interfering RNAs and small molecules represent promising steps towards disease-modifying therapies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
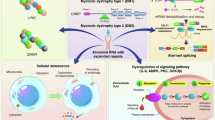

References
Brook, J. D. et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808 (1992).
Mahadevan, M. et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255, 1253–1255 (1992).
Liao, Q., Zhang, Y., He, J. & Huang, K. Global prevalence of myotonic dystrophy: an updated systematic review and meta-analysis. Neuroepidemiology 56, 163–173 (2022).
Johnson, N. E. et al. Population-based prevalence of myotonic dystrophy type 1 using genetic analysis of statewide blood screening program. Neurology 96, e1045–e1053 (2021).
Harper, P. S. Myotonic Dystrophy 3rd edn, 37 (WB Saunders, 2001).
Zapata-Aldana, E., Ceballos-Saenz, D., Hicks, R. & Campbell, C. Prenatal, neonatal, and early childhood features in congenital myotonic dystrophy. J. Neuromuscul. Dis. 5, 331–340 (2018).
Johnson, N. E. et al. Parent-reported multi-national study of the impact of congenital and childhood onset myotonic dystrophy. Dev. Med. Child. Neurol. 58, 698–705 (2016).
Lagrue, E. et al. A large multicenter study of pediatric myotonic dystrophy type 1 for evidence-based management. Neurology 92, e852–e865 (2019).
Johnson, N. E. et al. Disease burden and functional outcomes in congenital myotonic dystrophy: a cross-sectional study. Neurology 87, 160–167 (2016).
Campbell, C., Levin, S., Siu, V. M., Venance, S. & Jacob, P. Congenital myotonic dystrophy: Canadian population-based surveillance study. J. Pediatr. 163, 120–125 e121–e123 (2013).
Maagdenberg, S. J. M. et al. Impact of gastrointestinal and urological symptoms in children with myotonic dystrophy type 1. Neuromuscul. Disord. 35, 1–7 (2024).
Quigg, K. H. et al. 12-month progression of motor and functional outcomes in congenital myotonic dystrophy. Muscle Nerve 63, 384–391 (2021).
Reardon, W., Newcombe, R., Fenton, I., Sibert, J. & Harper, P. S. The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch. Dis. Child. 68, 177–181 (1993).
Aden, P., Skarbo, A. B., Wallace, S., Orstavik, K. & Rasmussen, M. Cognitive function, behaviour and quality of life in children with myotonic dystrophy type 1 in south-eastern Norway. Eur. J. Paediatr. Neurol. 45, 1–6 (2023).
Douniol, M. et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child. Neurol. 54, 905–911 (2012).
Logigian, E. L. et al. Quantitative analysis of the “warm-up” phenomenon in myotonic dystrophy type 1. Muscle Nerve 32, 35–42 (2005).
Okkersen, K. et al. The cognitive profile of myotonic dystrophy type 1: a systematic review and meta-analysis. Cortex 95, 143–155 (2017).
Okkersen, K. et al. Brain imaging in myotonic dystrophy type 1: a systematic review. Neurology 89, 960–969 (2017).
de Die-Smulders, C. E. et al. Age and causes of death in adult-onset myotonic dystrophy. Brain 121, 1557–1563 (1998).
Mathieu, J., Allard, P., Potvin, L. & Prévost, C. & Bégin, P. a 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 52, 1658–1662 (1999).
Johnson, N. E., Abbott, D. & Cannon-Albright, L. A. Relative risks for comorbidities associated with myotonic dystrophy: a population-based analysis. Muscle Nerve 52, 659–661 (2015).
Wahbi, K. et al. Development and validation of a new scoring system to predict survival in patients with myotonic dystrophy type 1. JAMA Neurol. 75, 573–581 (2018).
Gadalla, S. M. et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA 306, 2480–2486 (2011).
Emparanza, J. I. et al. Cancer phenotype in myotonic dystrophy patients: results from a meta-analysis. Muscle Nerve 58, 517–522 (2018).
Gadalla, S. M. et al. Quantifying cancer absolute risk and cancer mortality in the presence of competing events after a myotonic dystrophy diagnosis. PLoS ONE8, e79851 (2013).
Joosten, I. B. T. et al. Myotonic dystrophy type 1: a comparison between the adult- and late-onset subtype. Muscle Nerve 67, 130–137 (2023).
Harley, H. G. et al. Localisation of the myotonic dystrophy locus to 19q13.2–19q13.3 and its relationship to twelve polymorphic loci on 19q. Hum. Genet. 87, 73–80 (1991).
Harley, H. G. et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 355, 545–546 (1992).
Fu, Y. H. et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science 260, 235–238 (1993).
Jansen, G. et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 13, 316–324 (1996).
Berul, C. I. et al. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Invest. 103, R1–R7 (1999).
Frisch, R. et al. Effect of triplet repeat expansion on chromatin structure and expression of DMPK and neighboring genes, SIX5 and DMWD, in myotonic dystrophy. Mol. Genet. Metab. 74, 281–291 (2001).
Inukai, A. et al. Reduced expression of DMAHP/SIX5 gene in myotonic dystrophy muscle. Muscle Nerve 23, 1421–1426 (2000).
Yanovsky-Dagan, S. et al. Uncovering the role of hypermethylation by CTG expansion in myotonic dystrophy type 1 using mutant human embryonic stem cells. Stem Cell Rep. 5, 221–231 (2015).
Klesert, T. R. et al. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat. Genet. 25, 105–109 (2000).
Davis, B. M., McCurrach, M. E., Taneja, K. L., Singer, R. H. & Housman, D. E. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl Acad. Sci. USA 94, 7388–7393 (1997).
Jiang, H., Mankodi, A., Swanson, M. S., Moxley, R. T. & Thornton, C. A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13, 3079–3088 (2004).
Mankodi, A., Lin, X., Blaxall, B. C., Swanson, M. S. & Thornton, C. A. Nuclear RNA foci in the heart in myotonic dystrophy. Circ. Res. 97, 1152–1155 (2005).
Mankodi, A. et al. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol. 54, 760–768 (2003).
Mankodi, A. et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289, 1769–1773 (2000).
Napierała, M. & Krzyzosiak, W. J. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 272, 31079–31085 (1997).
Mooers, B. H. M., Logue, J. S. & Berglund, J. A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl Acad. Sci. USA 102, 16626–16631 (2005).
Hale, M. A., Johnson, N. E. & Berglund, J. A. Repeat-associated RNA structure and aberrant splicing. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 194405 (2019).
Ciesiolka, A., Jazurek, M., Drazkowska, K. & Krzyzosiak, W. J. Structural characteristics of simple RNA repeats associated with disease and their deleterious protein interactions. Front. Cell Neurosci. 11, 97 (2017).
Jain, A. & Vale, R. D. RNA phase transitions in repeat expansion disorders. Nature 546, 243 (2017).
Mankodi, A. et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 10, 2165–2170 (2001).
Miller, J. W. et al. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448 (2000).
Kanadia, R. N. et al. A muscleblind knockout model for myotonic dystrophy. Science 302, 1978–1980 (2003).
Lee, K.-Y. et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 5, 1887–1900 (2013).
Artero, R. et al. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol. 195, 131–143 (1998).
Oddo, J. C., Saxena, T., McConnell, O. L., Berglund, J. A. & Wang, E. T. Conservation of context-dependent splicing activity in distant muscleblind homologs. Nucleic Acids Res. 44, 8352–8362 (2016).
Wang, E. T. et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 25, 858–871 (2015).
Kalsotra, A. et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl Acad. Sci. USA 105, 20333–20338 (2008).
Lin, X. et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 15, 2087–2097 (2006).
Charizanis, K. et al. Muscleblind-like 2 mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75, 437–450 (2012).
Weyn-Vanhentenryck, S. M. et al. Precise temporal regulation of alternative splicing during neural development. Nat. Commun. 9, 2189 (2018).
Fardaei, M. et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 11, 805–814 (2002).
Kanadia, R. N. et al. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr. Patterns 3, 459–462 (2003).
Thomas, J. D. et al. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes. Dev. 31, 1122–1133 (2017).
Spruce, T. et al. The X-linked splicing regulator MBNL3 has been co-opted to restrict placental growth in eutherians. PLoS Biol. 20, e3001615 (2022).
Hildebrandt, R. P. et al. Muscleblind-like proteins use modular domains to localize RNAs by riding kinesins and docking to membranes. Nat. Commun. 14, 3427 (2023).
Wang, E. T. et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150, 710–724 (2012).
Masuda, A. et al. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep. 2, 209 (2012).
Du, H. et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 17, 187–193 (2010).
Osborne, R. J. et al. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 18, 1471–1481 (2009).
Batra, R. et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 56, 311–322 (2014).
Rau, F. et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 18, 840–845 (2011).
Piasecka, A. et al. MBNL splicing factors regulate the microtranscriptome of skeletal muscles. Nucleic Acids Res. 52, 12055–12073 (2024).
Goers, E. S., Purcell, J., Voelker, R. B., Gates, D. P. & Berglund, J. A. MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res. 38, 2467–2484 (2010).
Querido, E., Gallardo, F., Beaudoin, M., Ménard, C. & Chartrand, P. Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell Sci. 124, 1703–1714 (2011).
Sznajder, J. et al. Mechanistic determinants of MBNL activity. Nucleic Acids Res. 44, 10326–10342 (2016).
Kanadia, R. N. et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl Acad. Sci. USA 103, 11748–11753 (2006).
Dasgupta, T. & Ladd, A. N. The importance of CELF control: molecular and biological roles of the CUG-BP, elav-like family of RNA binding proteins. Wiley Interdisc. Rev. RNA 3, 104–121 (2012).
Shi, D.-L. & Grifone, R. RNA-binding proteins in the post-transcriptional control of skeletal muscle development, regeneration and disease. Front. Cell Dev. Biol. 9, 738978 (2021).
Peng, S. et al. The role of CELF family in neurodevelopment and neurodevelopmental disorders. Neurobiol. Dis. 197, 106525 (2024).
Fardaei, M., Larkin, K., Brook, J. D. & Hamshere, M. G. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 29, 2766–2771 (2001).
Savkur, R. S., Philips, A. V. & Cooper, T. A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29, 40–47 (2001).
Kuyumcu-Martinez, N. M., Wang, G.-S. & Cooper, T. A. Increased steady state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyper-phosphorylation. Mol. Cell 28, 68–78 (2007).
Ward, A. J., Rimer, M., Killian, J. M., Dowling, J. J. & Cooper, T. A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 19, 3614–3622 (2010).
Timchenko, N. A. et al. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J. Biol. Chem. 279, 13129–13139 (2004).
Ho, T. H., Bundman, D., Armstrong, D. L. & Cooper, T. A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 14, 1539–1547 (2005).
Wang, G.-S., Kearney, D. L., De Biasi, M., Taffet, G. & Cooper, T. A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Invest. 117, 2802–2811 (2007).
Kalsotra, A., Wang, K., Li, P.-F. & Cooper, T. A. MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes. Dev. 24, 653–658 (2010).
Kalsotra, A. et al. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue dramatically altering miRNA and mRNA expression. Cell Rep. 6, 336–345 (2014).
Ladd, A. N., Stenberg, M. G., Swanson, M. S. & Cooper, T. A. Dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Dev. Dyn. 233, 783–793 (2005).
Ladd, A. N., Charlet-B, N. & Cooper, T. A. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell Biol. 21, 1285–1296 (2001).
Koshelev, M., Sarma, S., Price, R. E., Wehrens, X. H. T. & Cooper, T. A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 19, 1066–1075 (2010).
Baralle, F. E. & Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451 (2017).
Kalsotra, A. & Cooper, T. A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729 (2011).
Wang, Z. & Burge, C. B. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813 (2008).
Otero, B. A. et al. Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep. 34, 108634 (2021).
Wang, E. T. et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 28, 1312–1321 (2019).
Wagner, S. D. et al. Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet. 12, e1006316 (2016).
Hale, M. A., Bates, K., Provenzano, M. & Johnson, N. E. Dynamics and variability of transcriptomic dysregulation in congenital myotonic dystrophy during pediatric development. Hum. Mol. Genet. 32, 1413–1428 (2022).
Degener, M. J. F. et al. A comprehensive atlas of fetal splicing patterns in the brain of adult myotonic dystrophy type 1 patients. Nar. Genom. Bioinform. 4, lqac016 (2022).
Thomas, J. D., Oliveira, R., Sznajder, J. & Swanson, M. S. Myotonic dystrophy and developmental regulation of RNA processing. Compr. Physiol. 8, 509–553 (2018).
López-Martínez, A., Soblechero-Martín, P., de-la-Puente-Ovejero, L., Nogales-Gadea, G. & Arechavala-Gomeza, V. An overview of alternative splicing defects implicated in myotonic dystrophy type I. Genes 11, 1109 (2020).
Lueck, J. D. et al. Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am. J. Physiol. Cell Physiol. 292, C1291–C1297 (2007).
Mankodi, A. et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 10, 35–44 (2002).
Kino, Y. et al. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res. 37, 6477–6490 (2009).
Cannon, S. C. Channelopathies of skeletal muscle excitability. Compr. Physiol. 5, 761–790 (2015).
Pedersen, T. H., Riisager, A., de Paoli, F. V., Chen, T.-Y. & Nielsen, O. B. Role of physiological ClC-1 Cl− ion channel regulation for the excitability and function of working skeletal muscle. J. Gen. Physiol. 147, 291–308 (2016).
Charlet-B, N. et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 10, 45–53 (2002).
Lueck, J. D., Mankodi, A., Swanson, M. S., Thornton, C. A. & Dirksen, R. T. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J. Gen. Physiol. 129, 79–94 (2007).
Wheeler, T. M., Lueck, J. D., Swanson, M. S., Dirksen, R. T. & Thornton, C. A. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Investig. 117, 3952–3957 (2007).
Koebis, M. et al. Ultrasound-enhanced delivery of morpholino with bubble liposomes ameliorates the myotonia of myotonic dystrophy model mice. Sci. Rep. 3, 2242 (2013).
Tuluc, P. et al. A CaV1.1 Ca2+ channel splice variant with high conductance and voltage-sensitivity alters EC coupling in developing skeletal muscle. Biophys. J. 96, 35–44 (2009).
Tang, Z. Z. et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of CaV1.1 calcium channel. Hum. Mol. Genet. 21, 1312–1324 (2012).
Cisco, L. A., Sipple, M. T., Edwards, K. M., Thornton, C. A. & Lueck, J. D. Verapamil mitigates chloride and calcium bi-channelopathy in a myotonic dystrophy mouse model. J. Clin. Investig. 134, e173576 (2024).
Nakamori, M. et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 74, 862–872 (2013).
Hartman, J. M. et al. RNA mis-splicing in children with congenital myotonic dystrophy is associated with physical function. Ann. Clin. Transl. Neurol. 11, 3175–3191 (2024).
Freyermuth, F. et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun. 7, 11067 (2016).
Pang, P. D. et al. CRISPR-mediated expression of the fetal Scn5a isoform in adult mice causes conduction defects and arrhythmias. J. Am. Heart Assoc. 7, e010393 (2018).
Wahbi, K. et al. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch. Cardiovasc. Dis. 106, 635–643 (2013).
Onkal, R. et al. Alternative splicing of Nav1.5: an electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms and critical involvement of a lysine residue. J. Cell Physiol. 216, 716–726 (2008).
Nitschke, L., Hu, R.-C., Miller, A. N. & Cooper, T. A. Rescue of Scn5a mis-splicing does not improve the structural and functional heart defects of a DM1 heart mouse model. Hum. Mol. Genet. 33, 1789–1799 (2024).
Seino, S. & Bell, G. I. Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 159, 312–316 (1989).
Moller, D. E., Yokota, A., Caro, J. F. & Flier, J. S. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 3, 1263–1269 (1989).
Belfiore, A. et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr. Rev. 38, 379–431 (2017).
Kellerer, M. et al. Distinct alpha-subunit structures of human insulin receptor A and B variants determine differences in tyrosine kinase activities. Biochemistry 31, 4588–4596 (1992).
Ho, T. H. et al. Muscleblind proteins regulate alternative splicing. EMBO J. 23, 3103–3112 (2004).
Sen, S. et al. Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem. 285, 25426–25437 (2010).
Echeverria, G. V. & Cooper, T. A. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 42, 1893–1903 (2014).
Dahlqvist, J. R. & Vissing, J. in Diabetes Associated with Single Gene Defects and Chromosomal Abnormalities (eds Barbetti, F. et al.) 182–187 (Karger, 2017).
Moxley, R. T., Corbett, A. J., Minaker, K. L. & Rowe, J. W. Whole body insulin resistance in myotonic dystrophy. Ann. Neurol. 15, 157–162 (1984).
Vialettes, B. et al. Mechanism and significance of insulin resistance in myotonic dystrophy. Horm. Metab. Res. 18, 395–399 (1986).
Goodwin, M. et al. MBNL sequestration by toxic RNAs and RNA mis-processing in the myotonic dystrophy brain. Cell Rep. 12, 1159–1168 (2015).
Barbier, P. et al. Role of tau as a microtubule-associated protein: structural and functional aspects. Front. Aging Neurosci. 11, 204 (2019).
Dhaenens, C. M. et al. Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: two individual consequences of CUG trinucleotide repeats. Exp. Neurol. 210, 467–478 (2008).
Hefti, M. M. et al. High-resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS ONE 13, e0195771 (2018).
Carpentier, C. et al. Tau exon 2 responsive elements deregulated in myotonic dystrophy type I are proximal to exon 2 and synergistically regulated by MBNL1 and MBNL2. Biochim. Biophys. Acta 1842, 654–664 (2014).
Dhaenens, C. M. et al. Mis-splicing of Tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim. Biophys. Acta 1812, 732–742 (2011).
Rawat, P. et al. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int. J. Mol. Sci. 23, 12841 (2022).
Weijs, R. et al. Human brain pathology in myotonic dystrophy type 1: a systematic review. Neuropathology 41, 3–20 (2021).
Vermersch, P. et al. Specific tau variants in the brains of patients with myotonic dystrophy. Neurology 47, 711–717 (1996).
Hamasaki, H. et al. Neuropathology of classic myotonic dystrophy type 1 is characterized by both early initiation of primary age-related tauopathy of the hippocampus and unique 3-repeat tauopathy of the brainstem. J. Neuropathol. Exp. Neurol. 82, 29–37 (2022).
Sznajder, L. J. et al. Autism-related traits in myotonic dystrophy type 1 model mice are due to MBNL sequestration and RNA mis-splicing of autism-risk genes. Nat. Neurosci. 28, 1199–1212 (2025).
Koehorst, E., Ballester-Lopez, A., Arechavala-Gomeza, V., Martínez-Piñeiro, A. & Nogales-Gadea, G. The biomarker potential of miRNAs in myotonic dystrophy type I. J. Clin. Med. 9, 3939 (2020).
Voellenkle, C. et al. Dysregulation of circular RNAs in myotonic dystrophy type 1. Int. J. Mol. Sci. 20, 1938 (2019).
Czubak, K. et al. Global increase in circular RNA levels in myotonic dystrophy. Front. Genet. 10, 649 (2019).
Czubak, K., Sedehizadeh, S., Kozlowski, P. & Wojciechowska, M. An overview of circular RNAs and their implications in myotonic dystrophy. Int. J. Mol. Sci. 20, 4385 (2019).
Zu, T., Pattamatta, A. & Ranum, L. P. W. Repeat-associated non-ATG translation in neurological diseases. Cold Spring Harb. Persp. Biol. 10, a033019 (2018).
Fujino, Y., Mori, K. & Nagai, Y. Repeat-associated non-AUG translation in neuromuscular diseases: mechanisms and therapeutic insights. J. Biochem. 173, 273–281 (2023).
Zu, T. et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA 108, 260–265 (2011).
Zu, T. et al. RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 95, 1292–1305.e1295 (2017).
Koehorst, E. et al. Characterization of RAN translation and antisense transcription in primary cell cultures of patients with myotonic dystrophy type 1. J. Clin. Med. 10, 5520 (2021).
Morales, F. et al. Longitudinal increases in somatic mosaicism of the expanded CTG repeat in myotonic dystrophy type 1 are associated with variation in age-at-onset. Hum. Mol. Genet. 29, 2496–2507 (2020).
Overend, G. et al. Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum. Mol. Genet. 28, 2245–2254 (2019).
Hogrel, J. Y. et al. Relationships between grip strength, myotonia, and CTG expansion in myotonic dystrophy type 1. Ann. Clin. Transl. Neurol. 4, 921–925 (2017).
Higham, C. F., Morales, F., Cobbold, C. A., Haydon, D. T. & Monckton, D. G. High levels of somatic DNA diversity at the myotonic dystrophy type 1 locus are driven by ultra-frequent expansion and contraction mutations. Hum. Mol. Genet. 21, 2450–2463 (2012).
Morales, F. et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet. 21, 3558–3567 (2012).
Higham, C. F. & Monckton, D. G. Modelling and inference reveal nonlinear length-dependent suppression of somatic instability for small disease associated alleles in myotonic dystrophy type 1 and Huntington disease. J. R. Soc. Interf.10, 20130605 (2013).
Nakamori, M. et al. Cell type-specific abnormalities of central nervous system in myotonic dystrophy type 1. Brain Commun. 4, fcac154 (2022).
Thornton, C. A., Johnson, K. & Moxley, R. T. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol. 35, 104–107 (1994).
Morales, F. et al. Individual-specific levels of CTG•CAG somatic instability are shared across multiple tissues in myotonic dystrophy type 1. Hum. Mol. Genet. 32, 621–631 (2023).
Monckton, D. G., Wong, L. J., Ashizawa, T. & Caskey, C. T. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum. Mol. Genet. 4, 1–8 (1995).
Barbé, L. et al. CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am. J. Hum. Genet. 100, 488–505 (2017).
Pearson, C. E., Edamura, N. K. & Cleary, J. D. Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 6, 729–742 (2005).
Yum, K., Wang, E. T. & Kalsotra, A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr. Opin. Genet. Dev. 44, 30–37 (2017).
Gacy, A. M., Goellner, G., Juranić, N., Macura, S. & McMurray, C. T. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81, 533–540 (1995).
Axford, M. M. et al. Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLoS Genet. 9, e1003866 (2013).
Tam, M. et al. Slipped (CTG)·(CAG) repeats of the myotonic dystrophy locus: surface probing with anti-DNA antibodies. J. Mol. Biol. 332, 585–600 (2003).
Salinas-Rios, V., Belotserkovskii, B. P. & Hanawalt, P. C. DNA slip-outs cause RNA polymerase II arrest in vitro: potential implications for genetic instability. Nucleic Acids Res. 39, 7444–7454 (2011).
Reddy, K. et al. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 39, 1749–1762 (2011).
Reddy, K. et al. Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Res. 42, 10473–10487 (2014).
Lin, Y., Dent, S. Y. R., Wilson, J. H., Wells, R. D. & Napierala, M. R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl Acad. Sci. USA 107, 692–697 (2010).
Morales, F. et al. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair. 40, 57–66 (2016).
Flower, M. et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain142, 1876–1886 (2019).
Foiry, L. et al. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum. Genet. 119, 520–526 (2006).
van den Broek, W. J. A. A. et al. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet. 11, 191–198 (2002).
Guo, J., Gu, L., Leffak, M. & Li, G.-M. MutSβ promotes trinucleotide repeat expansion by recruiting DNA polymerase β to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res. 26, 775–786 (2016).
Pearson, C. E., Ewel, A., Acharya, S., Fishel, R. A. & Sinden, R. R. Human MSH2 binds to trinucleotide repeat DNA structures associated with neurodegenerative diseases. Hum. Mol. Genet. 6, 1117–1123 (1997).
López Castel, A., Cleary, J. D. & Pearson, C. E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 11, 165–170 (2010).
O’Reilly, D. et al. Di-valent siRNA-mediated silencing of MSH3 blocks somatic repeat expansion in mouse models of Huntington’s disease. Mol. Ther. 31, 1661–1674 (2023).
Porro, A. et al. FAN1–MLH1 interaction affects repair of DNA interstrand cross-links and slipped-CAG/CTG repeats. Sci. Adv. 7, eabf7906 (2021).
Musova, Z. et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. A 149A, 1365–1374 (2009).
Peric, S., Pesovic, J., Savic-Pavicevic, D., Rakocevic Stojanovic, V. & Meola, G. Molecular and clinical implications of variant repeats in myotonic dystrophy type 1. Int. J. Mol. Sci. 23, 354 (2021).
Braida, C. et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 19, 1399–1412 (2010).
Botta, A. et al. Identification and characterization of 5′ CCG interruptions in complex DMPK expanded alleles. Eur. J. Hum. Genet. 25, 257–261 (2017).
Cumming, S. A. et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur. J. Hum. Genet. 26, 1635–1647 (2018).
Miller, J. N. et al. Variant repeats within the DMPK CTG expansion protect function in myotonic dystrophy type 1. Neurol. Genet. 6, e504 (2020).
Pešović, J. et al. Repeat interruptions modify age at onset in myotonic dystrophy type 1 by stabilizing DMPK expansions in somatic cells. Front. Genet. 9, 601 (2018).
Wenninger, S. et al. Associations between variant repeat interruptions and clinical outcomes in myotonic dystrophy type 1. Neurol. Genet. 7, e572 (2021).
Buckley, L., Lacey, M. & Ehrlich, M. Epigenetics of the myotonic dystrophy-associated DMPK gene neighborhood. Epigenomics 8, 13–31 (2016).
López Castel, A. et al. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet. 20, 1–15 (2011).
Morales, F. et al. Myotonic dystrophy type 1 (DM1) clinical subtypes and CTCF site methylation status flanking the CTG expansion are mutant allele length-dependent. Hum. Mol. Genet. 31, 262–274 (2021).
de Pontual, L. & Tomé, S. Overview of the complex relationship between epigenetics markers, CTG repeat instability and symptoms in myotonic dystrophy type 1. Int. J. Mol. Sci. 23, 3477 (2022).
Visconti, V. V. et al. Epigenetics of myotonic dystrophies: a minireview. Int. J. Mol. Sci. 22, 12594 (2021).
Breton, É. et al. DNA methylation at the DMPK gene locus is associated with cognitive functions in myotonic dystrophy type 1. Epigenomics 12, 2051–2064 (2020).
Légaré, C. et al. DMPK gene DNA methylation levels are associated with muscular and respiratory profiles in DM1. Neurol. Genet. 5, e338 (2019).
Izzo, M. et al. Molecular therapies for myotonic dystrophy type 1: from small drugs to gene editing. Int. J. Mol. Sci. 23, 4622 (2022).
Ashizawa, T. et al. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurol. Clin. Pract. 8, 507–520 (2018).
McNally, E. M. et al. Clinical care recommendations for cardiologists treating adults with myotonic dystrophy. J. Am. Heart Assoc. 9, e014006 (2020).
Johnson, N. E. et al. Consensus-based care recommendations for congenital and childhood-onset myotonic dystrophy type 1. Neurol. Clin. Pract. 9, 443–454 (2019).
Boentert, M. et al. Consensus-based care recommendations for pulmonologists treating adults with myotonic dystrophy type 1. Respiration 99, 360–368 (2020).
Veyckemans, F. & Scholtes, J. L. Myotonic dystrophies type 1 and 2: anesthetic care. Paediat. Anaesth. 23, 794–803 (2013).
Hartman, J., Patki, T. & Johnson, N. E. Diagnosis and management of myotonic dystrophy type 1. JAMA 331, 1227–1228 (2024).
Heatwole, C. et al. Mexiletine in myotonic dystrophy type 1: a randomized, double-blind, placebo-controlled trial. Neurology 96, e228–e240 (2021).
Orlikowski, D. et al. Modafinil for the treatment of hypersomnia associated with myotonic muscular dystrophy in adults: a multicenter, prospective, randomized, double-blind, placebo-controlled, 4-week trial. Clin. Ther. 31, 1765–1773 (2009).
Mikhail, A. I. et al. Aerobic exercise elicits clinical adaptations in myotonic dystrophy type 1 patients independently of pathophysiological changes. J. Clin. Invest. 132, e156125 (2022).
Orngreen, M. C., Olsen, D. B. & Vissing, J. Aerobic training in patients with myotonic dystrophy type 1. Ann. Neurol. 57, 754–757 (2005).
Brady, L. I., MacNeil, L. G. & Tarnopolsky, M. A. Impact of habitual exercise on the strength of individuals with myotonic dystrophy type 1. Am. J. Phys. Med. Rehabil. 93, 739–746 (2014).
Gallais, B., Roussel, M. P., Laberge, L., Hebert, L. J. & Duchesne, E. Impact of a 12-week strength training program on fatigue, daytime sleepiness, and apathy in men with myotonic dystrophy type 1. J. Neuromuscul. Dis. 9, 629–639 (2022).
Girard-Cote, L. et al. Resistance training in women with myotonic dystrophy type 1: a multisystemic therapeutic avenue. Neuromuscul. Disord. 40, 38–51 (2024).
Okkersen, K. et al. Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type 1: a multicentre, single-blind, randomised trial. Lancet Neurol. 17, 671–680 (2018).
Horrigan, J. et al. A phase 2 study of AMO-02 (tideglusib) in congenital and childhood-onset myotonic dystrophy type 1 (DM1). Pediatr. Neurol. 112, 84–93 (2020).
Nakamori, M. et al. Erythromycin for myotonic dystrophy type 1: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. EClinicalMedicine 67, 102390 (2024).
Bassez, G. et al. Improved mobility with metformin in patients with myotonic dystrophy type 1: a randomized controlled trial. Brain 141, 2855–2865 (2018).
Avidity Biosciences announces new positive AOC 1001 data demonstrating improvement in multiple additional functional endpoints and favorable long-term safety and tolerability in people with myotonic dystrophy type 1. Avidity Biosciences https://aviditybiosciences.investorroom.com/2023-10-07-Avidity-Biosciences-Announces-New-Positive-AOC-1001-Data-Demonstrating-Improvement-in-Multiple-Additional-Functional-Endpoints-and-Favorable-Long-term-Safety-and-Tolerability-in-People-with-Myotonic-Dystrophy-Type-1 (2023).
Dyne Therapeutics announces new clinical data from ACHIEVE trial of DYNE-101 in DM1 and DELIVER trial of DYNE-251 in DMD demonstrating compelling impact on key disease biomarkers and improvement in multiple functional endpoints. Dyne Therapeutics https://investors.dyne-tx.com/news-releases/news-release-details/dyne-therapeutics-announces-new-clinical-data-achieve-trial-dyne/ (2024).
Pascual-Gilabert, M., Artero, R. & Lopez-Castel, A. The myotonic dystrophy type 1 drug development pipeline: 2022 edition. Drug. Discov. Today 28, 103489 (2023).
Timchenko, L. Development of therapeutic approaches for myotonic dystrophies type 1 and type 2. Int. J. Mol. Sci. 23, 10491 (2022).
Wang, M. et al. Correction of glycogen synthase kinase 3β in myotonic dystrophy 1 reduces the mutant RNA and improves postnatal survival of DMSXL mice. Mol. Cell. Biol. 39, e00155-19 (2019).
Garcia-Puga, M., Saenz-Antonanzas, A., Matheu, A. & Lopez de Munain, A. Targeting myotonic dystrophy type 1 with metformin. Int. J. Mol. Sci. 23, 2901 (2022).
Repellin, M. et al. Repurposing pentamidine using hyaluronic acid-based nanocarriers for skeletal muscle treatment in myotonic dystrophy. Nanomedicine 47, 102623 (2023).
Corum, D. G. et al. PDE5 inhibition rescues mitochondrial dysfunction and angiogenic responses induced by Akt3 inhibition by promotion of PRC expression. J. Biol. Chem. 295, 18091–18104 (2020).
Reddy, K., Jenquin, J. R., Cleary, J. D. & Berglund, J. A. Mitigating RNA toxicity in myotonic dystrophy using small molecules. Int. J. Mol. Sci. 20, 4017 (2019).
Rinaldi, C. & Wood, M. J. A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21 (2018).
El Boujnouni, N. et al. Block or degrade? Balancing on- and off-target effects of antisense strategies against transcripts with expanded triplet repeats in DM1. Mol. Ther. Nucl. Acids 32, 622–636 (2023).
Pascual-Gilabert, M., Lopez-Castel, A. & Artero, R. Myotonic dystrophy type 1 drug development: a pipeline toward the market. Drug. Discov. Today 26, 1765–1772 (2021).
Cerro-Herreros, E. et al. Therapeutic potential of antagomiR-23b for treating myotonic dystrophy. Mol. Ther. Nucl. Acids 21, 837–849 (2020).
Cerro-Herreros, E. et al. miR-23b and miR-218 silencing increase muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 9, 2482 (2018).
Cerro-Herreros, E. et al. Preclinical characterization of antagomiR-218 as a potential treatment for myotonic dystrophy. Mol. Ther. Nucl. Acids 26, 174–191 (2021).
Tedesco, F. S. et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 4, 140ra189 (2012).
Gerli, M. F. M., Maffioletti, S. M., Millet, Q. & Tedesco, F. S. Transplantation of induced pluripotent stem cell-derived mesoangioblast-like myogenic progenitors in mouse models of muscle regeneration. J. Vis. Exp. 20, e50532 (2014).
Raaijmakers, R. H. L. et al. Ameliorated cellular hallmarks of myotonic dystrophy in hybrid myotubes from patient and unaffected donor cells. Stem Cell Res. Ther. 15, 302 (2024).
Sampaolesi, M. et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444, 574–579 (2006).
Porquet, F. et al. Specific-promoter targeting by CRISPRi reverses myotonic dystrophy type 1-associated defects in patient muscle cells. Mol. Ther. Nucl. Acids 32, 857–871 (2023).
Raaijmakers, R. H. L., Ripken, L., Ausems, C. R. M. & Wansink, D. G. CRISPR/Cas applications in myotonic dystrophy: expanding opportunities. Int. J. Mol. Sci. 20, 3689 (2019).
Marsh, S., Hanson, B., Wood, M. J. A., Varela, M. A. & Roberts, T. C. Application of CRISPR–Cas9-mediated genome editing for the treatment of myotonic dystrophy type 1. Mol. Ther. 28, 2527–2539 (2020).
van Agtmaal, E. L. et al. CRISPR/Cas9-induced (CTG⋅CAG)(n) repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther. 25, 24–43 (2017).
Provenzano, C. et al. CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol. Ther. Nucleic Acids 9, 337–348 (2017).
Dastidar, S. et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 46, 8275–8298 (2018).
Wang, Y. et al. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 26, 2617–2630 (2018).
Chulanova, Y., Breier, D. & Peer, D. Delivery of genetic medicines for muscular dystrophies. Cell Rep. Med. 6, 101885 (2025).
Thornton, C. A. et al. Antisense oligonucleotide targeting DMPK in patients with myotonic dystrophy type 1: a multicentre, randomised, dose-escalation, placebo-controlled, phase 1/2a trial. Lancet Neurol. 22, 218–228 (2023).
Sedehizadeh, S., Wojciechowska, M., Ketley, A., Brook, J. D. & Maddison, P. Splicing in two skeletal muscle transcripts correlates with clinical phenotype in myotonic dystrophy type 1 patients. J. Neurol. 269, 2784–2787 (2022).
Provenzano, M. et al. The splice index as a prognostic biomarker of strength and function in myotonic dystrophy type 1. J. Clin. Invest. 135, e185426 (2025).
Antoury, L. et al. Analysis of extracellular mRNA in human urine reveals splice variant biomarkers of muscular dystrophies. Nat. Commun. 9, 3906 (2018).
van Cruchten, R. T. P. et al. Clinical improvement of DM1 patients reflected by reversal of disease-induced gene expression in blood. BMC Med. 20, 395 (2022).
Garibaldi, M. et al. Muscle magnetic resonance imaging in myotonic dystrophy type 1 (DM1): refining muscle involvement and implications for clinical trials. Eur. J. Neurol. 29, 843–854 (2022).
van der Plas, E. et al. Quantitative muscle MRI as a sensitive marker of early muscle pathology in myotonic dystrophy type 1. Muscle Nerve 63, 553–562 (2021).
Fionda, L. et al. Muscle MRI as a biomarker of disease activity and progression in myotonic dystrophy type 1: a longitudinal study. J. Neurol. 271, 5864–5874 (2024).
Heskamp, L. et al. Quantitative muscle MRI depicts increased muscle mass after a behavioral change in myotonic dystrophy type 1. Radiology 297, 132–142 (2020).
Hamilton, M. J. et al. Elevated plasma levels of cardiac troponin-I predict left ventricular systolic dysfunction in patients with myotonic dystrophy type 1: a multicentre cohort follow-up study. PLoS ONE12, e0174166 (2017).
Rossi, S. & Silvestri, G. Fluid biomarkers of central nervous system (CNS) involvement in myotonic dystrophy type 1 (DM1). Int. J. Mol. Sci. 24, 2204 (2023).
Tanner, M. K., Tang, Z. & Thornton, C. A. Targeted splice sequencing reveals RNA toxicity and therapeutic response in myotonic dystrophy. Nucleic Acids Res. 49, 2240–2254 (2021).
De Antonio, M. et al. The DM-scope registry: a rare disease innovative framework bridging the gap between research and medical care. Orphanet J. Rare Dis. 14, 122 (2019).
la Fontaine, L. A. et al. Comprehensive four-year disease progression assessment of myotonic dystrophy type 1. Neuromuscul. Disord. 43, 44–52 (2024).
Provenzano, M. et al. The splice index as a prognostic biomarker of strength and function in myotonic dystrophy type 1. J. Clin. Invest. 135, 4 (2025).
Mangin, A. et al. Robust detection of somatic mosaicism and repeat interruptions by long-read targeted sequencing in myotonic dystrophy type 1. Int. J. Mol. Sci. 22, 2616 (2021).
Estab biomarkers and clinical endpoints in myotonic dystrophy type 1 (END-DM1). Clinicaltrials.gov https://clinicaltrials.gov/study/NCT03981575 (2025).
Braz, S. O., Acquaire, J., Gourdon, G. & Gomes-Pereira, M. Of mice and men: advances in the understanding of neuromuscular aspects of myotonic dystrophy. Front. Neurol. 9, 519 (2018).
Neault, N. et al. Vorinostat improves myotonic dystrophy type 1 splicing abnormalities in DM1 muscle cell lines and skeletal muscle from a DM1 mouse model. Int. J. Mol. Sci. 24, 3794 (2023).
Huguet, A. et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 8, e1003043 (2012).
Seznec, H. et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet. 10, 2717–2726 (2001).
Decostre, V. et al. Longitudinal in vivo muscle function analysis of the DMSXL mouse model of myotonic dystrophy type 1. Neuromuscul. Disord. 23, 1016–1025 (2013).
Golini, E. et al. Excessive rest time during active phase is reliably detected in a mouse model of myotonic dystrophy type 1 using home cage monitoring. Front. Behav. Neurosci. 17, 1130055 (2023).
Potier, B. et al. DM1 transgenic mice exhibit abnormal neurotransmitter homeostasis and synaptic plasticity in association with RNA foci and mis-splicing in the hippocampus. Int. J. Mol. Sci. 23, 592 (2022).
Gomes-Pereira, M., Cooper, T. A. & Gourdon, G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol. Med. 17, 506–517 (2011).
Morriss, G. R., Rajapakshe, K., Huang, S., Coarfa, C. & Cooper, T. A. Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1. Hum. Mol. Genet. 27, 2789–2804 (2018).
Suenaga, K. et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS ONE 7, e33218 (2012).
Khandelwal, A. et al. Mbnl2 loss alters novel context processing and impairs object recognition memory. iScience 26, 106732 (2023).
Safety and efficacy of tideglusib in congenital or childhood onset myotonic dystrophy (REACH CDM X). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT05004129 (2023).
Efficacy and safety of tideglusib in congenital myotonic dystrophy. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT03692312 (2023).
AMO Pharma completes meeting with U.S. FDA and outlines plans to advance clinical development of AMO-02 (tideglusib) in treatment of myotonic dystrophy. AMO Pharma https://www.amo-pharma.com/news/Press_Release_240502.htm (2024).
Study of tideglusib in adolescent and adult patients with myotonic dystrophy. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT02858908 (2018).
A phase 2 blinded, placebo-controlled study to assess the safety, tolerability, and efficacy of MYD-0124 to adult patients with myotonic dystrophy type 1 — MYD-0124 study. World Health Organization https://trialsearch.who.int/?TrialID=JPRN-jRCT2051190069 (2024).
Efficacy of metformin on motility and strength in myotonic dystrophy type 1. EU Clinical Trials Register https://www.clinicaltrialsregister.eu/ctr-search/trial/2018-000692-32/IT (2020).
A randomized, double blind, placebo-controlled phase ii study of metformin in myotonic dystrophy type 1 patients. EU Clinical Trials Register https://www.clinicaltrialsregister.eu/ctr-search/search?query=2013-001732-21 (2013).
Global study of del-desiran for the treatment of DM1 (HARBOR). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT06411288 (2025).
Study of AOC 1001 in adult myotonic dystrophy type 1 (DM1) Patients (MARINA). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT05027269 (2024).
Safety, tolerability, pharmacodynamic, efficacy, and pharmacokinetic study of DYNE-101 in participants with myotonic dystrophy type 1 (ACHIEVE). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT05481879 (2025).
Study of ARO-DM1 in subjects with type 1 myotonic dystrophy. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT06138743 (2025).
Study of ATX-01 in participants with DM1 (ArthemiR). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT06300307 (2025).
Safety, tolerability, PK, and PD study of PGN-EDODM1 in participants with myotonic dystrophy type 1 (FREEDOM-DM1). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT06204809 (2025).
A phase 1/2 study of VX-670 in adult participants with myotonic dystrophy 1 (DM1) (Galileo). ClinicalTrials.gov https://clinicaltrials.gov/study/NCT06185764 (2025).
A safety andtolerability study of multiple doses of ISIS-DMPKRx in adults with myotonic dystrophy type 1. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT02312011 (2022).
Gambardella, S. et al. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J. Transl. Med. 8, 48 (2010).
Ambrose, K. K. et al. Deregulation of microRNAs in blood and skeletal muscles of myotonic dystrophy type 1 patients. Neurol. India 65, 512–517 (2017).
Fernandez-Costa, J. M. et al. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 22, 704–716 (2013).
Fritegotto, C., Ferrati, C., Pegoraro, V. & Angelini, C. Micro-RNA expression in muscle and fiber morphometry in myotonic dystrophy type 1. Neurol. Sci. 38, 619–625 (2017).
Perbellini, R. et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord.21, 81–88 (2011).
Aoussim, A. et al. Towards the identification of biomarkers for muscle function improvement in myotonic dystrophy type 1. J. Neuromuscul. Dis. 10, 1041–1053 (2023).
Varga, D. et al. Urinary titin in myotonic dystrophy type 1. Muscle Nerve 68, 215–218 (2023).
Nguyen, C. D. L. et al. Periostin as a blood biomarker of muscle cell fibrosis, cardiomyopathy and disease severity in myotonic dystrophy type 1. J. Neurol. 270, 3138–3158 (2023).
Perfetti, A. et al. Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul. Disord.24, 509–515 (2014).
Perfetti, A. et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 6, 38174 (2016).
Saak, A. et al. Serum neurofilament light chain: a marker of nervous system damage in myopathies. Front. Neurosci. 15, 791670 (2021).
van der Plas, E. et al. Blood-based markers of neuronal injury in adult-onset myotonic dystrophy type 1. Front. Neurol. 12, 791065 (2022).
Nicoletti, T. F. et al. Elevated serum neurofilament light chain (NfL) as a potential biomarker of neurological involvement in myotonic dystrophy type 1 (DM1). J. Neurol. 269, 5085–5092 (2022).
Winblad, S. et al. Cerebrospinal fluid tau and amyloid beta42 protein in patients with myotonic dystrophy type 1. Eur. J. Neurol. 15, 947–952 (2008).
Peric, S. et al. Cerebrospinal fluid biomarkers of neurodegeneration in patients with juvenile and classic myotonic dystrophy type 1. Eur. J. Neurol. 21, 231–237 (2014).
Laforce, R. et al. Tau positron emission tomography, cerebrospinal fluid and plasma biomarkers of neurodegeneration, and neurocognitive testing: an exploratory study of participants with myotonic dystrophy type 1. J. Neurol. 269, 3579–3587 (2022).
Conforti, R. et al. Brain MRI abnormalities in the adult form of myotonic dystrophy type 1: a longitudinal case series study. Neuroradiol. J. 29, 36–45 (2016).
Cabada, T. et al. Longitudinal study in patients with myotonic dystrophy type 1: correlation of brain MRI abnormalities with cognitive performances. Neuroradiology 63, 1019–1029 (2021).
Labayru, G. et al. Neurodegeneration trajectory in pediatric and adult/late DM1: a follow-up MRI study across a decade. Ann. Clin. Transl. Neurol. 7, 1802–1815 (2020).
Gliem, C. et al. Tracking the brain in myotonic dystrophies: a 5-year longitudinal follow-up study. PLoS ONE 14, e0213381 (2019).
Labayru, G. et al. White matter integrity changes and neurocognitive functioning in adult-late onset DM1: a follow-up DTI study. Sci. Rep. 12, 3988 (2022).
Minnerop, M., Gliem, C. & Kornblum, C. Current progress in CNS imaging of myotonic dystrophy. Front. Neurol. 9, 646 (2018).
Cerro-Herreros, E., Chakraborty, M., Perez-Alonso, M., Artero, R. & Llamusi, B. Expanded CCUG repeat RNA expression in Drosophila heart and muscle trigger myotonic dystrophy type 1-like phenotypes and activate autophagocytosis genes. Sci. Rep. 7, 2843 (2017).
deLorimier, E. et al. Modifications to toxic CUG RNAs induce structural stability, rescue mis-splicing in a myotonic dystrophy cell model and reduce toxicity in a myotonic dystrophy zebrafish model. Nucleic Acids Res. 42, 12768–12778 (2014).
Berenger-Currias, N., Martinat, C. & Baghdoyan, S. Pluripotent stem cells in disease modeling and drug discovery for myotonic dystrophy type 1. Cells 12, 571 (2023).
Ausems, C. R. M. et al. Intrinsic myogenic potential of skeletal muscle-derived pericytes from patients with myotonic dystrophy type 1. Mol. Ther. Meth. Clin. Dev. 15, 120–132 (2019).
Thornell, L. E. et al. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol. Appl. Neurobiol. 35, 603–613 (2009).
Bigot, A. et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am. J. Pathol. 174, 1435–1442 (2009).
Matloka, M., Klein, A. F., Rau, F. & Furling, D. Cells of matter-ml models for myotonic dystrophy. Front. Neurol. 9, 361 (2018).
Furling, D., Lemieux, D., Taneja, K. & Puymirat, J. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul. Disord. 11, 728–735 (2001).
Birbrair, A. & Delbono, O. Pericytes are essential for skeletal muscle formation. Stem Cell Rev. Rep. 11, 547–548 (2015).
Dellavalle, A. et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2, 499 (2011).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Poulin, H. et al. iPSC-derived cardiomyocytes from patients with myotonic dystrophy type 1 have abnormal ion channel functions and slower conduction velocities. Sci. Rep. 11, 2500 (2021).
Ueki, J. et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep. 7, 42522 (2017).
Frega, M. et al. Rapid neuronal differentiation of induced pluripotent stem cells for measuring network activity on micro-electrode arrays. J. Vis. Exp. 8, 54900 (2017).
Morelli, K. H. et al. MECP2-related pathways are dysregulated in a cortical organoid model of myotonic dystrophy. Sci. Transl. Med. 14, eabn2375 (2022).
Acknowledgements
Several authors of this publication are members of the Radboudumc Center of Expertise for Neuromuscular Disorders (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for Rare Neuromuscular Diseases (EURO-NMD).
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to discussion of the content. K.M., L.R. and M.A.H. wrote the article. R.H.L.R., A.M.Q., T.P., N.E.J. and H.v.B. reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
K.M. served as a paid consultant for Dyne Therapeutics and Avidity Biosciences (payments to institution). She receives research funding from PepGen Inc. N.E.J. has received grant funding from NINDS (R01NS104010, U01NS124974), NCATS (R21TR003184), the CDC (U01DD001242) and the FDA (7R01FD006071). N.E.J. receives royalties from the CCMDHI and the CMTHI. He receives research funds from Novartis, Takeda, PepGen, Sanofi Genzyme, Dyne, Vertex Pharmaceuticals, Fulcrum Therapeutics, AskBio, ML Bio, and Sarepta. N.E.J. has consulted for Arthex, Angle Therapeutics, Juvena, Rgenta, PepGen, AMO Pharma, Takeda, Design, Dyne, AskBio, Avidity and Vertex Pharmaceuticals. He has equity in Repeat RNA Therapuetics, Evolyra Therapeutics, and Juvena Therapeutics. M.A.H. has consulted for Juvena Therapeutics, Arrakis Therapeutics and Vertex Pharmaceuticals and retains equity in Evolyra Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Neurology thanks Benedikt Schoser and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rahm, L., Hale, M.A., Raaijmakers, R.H.L. et al. Myotonic dystrophy type 1: clinical diversity, molecular insights and therapeutic perspectives. Nat Rev Neurol 21, 623–641 (2025). https://doi.org/10.1038/s41582-025-01139-x
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41582-025-01139-x
