
Abstract
Background
Desmoglein-2 (DSG2) is an essential cardiac desmosomal cadherin, and its alteration underlies a broad spectrum of arrhythmogenic cardiomyopathy (ACM). Yet, the clinical significance of many DSG2 variants remains uncertain. This study aimed to systematically characterize the spectrum, structural impact, and clinical relevance of DSG2 variants by integrating large-scale genomic evidence, published data, and a deeply phenotyped validation cohort.
Methods
We conducted a systematic literature review (115 studies; 145 curated variants) and analyzed population-scale datasets (3570 variants in gnomAD; 1847 in ClinVar). All variants were uniformly reclassified following ACMG/ClinGen criteria. A validation cohort of 95 Italian DSG2-carriers underwent detailed phenotyping. Structural modeling via AlphaFold, supported protein modeling, calcium-binding site prediction, and DynaMut stability analysis were performed to evaluate the functional consequences of key variants.
Results
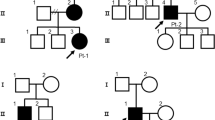
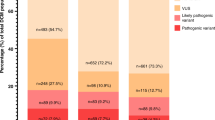
Literature and database integration reveal domain-specific variant clustering, with high-impact missense variants enriched in calcium-binding extracellular domains, the furin cleavage site, and the intracellular PKP2-binding region. In the validation cohort, penetrance among genotype-positive relatives is 42%, while 13% of definite ACM cases experience major ventricular arrhythmias; transplantation and mortality each occur in 3%. Biallelic and digenic variants are associated with earlier onset and more severe biventricular involvement. Structural modeling confirms that pathogenic missense substitutions destabilize DSG2 architecture or impair calcium-dependent adhesion.
Conclusions
This study refines the classification of DSG2 variants and highlights the importance of domain-level and multilocus interpretation in ACM. These findings support comprehensive genetic screening, structural modeling for variant assessment, and lifelong follow-up of DSG2 carriers to improve diagnosis and risk stratification.
Plain language summary
Arrhythmogenic cardiomyopathy (ACM) is an inherited heart disease that can cause abnormal rhythms and sudden cardiac death, often in young adults. This study focused on the DSG2 gene, which helps heart cells stick together. We combined genetic data from large population databases, published studies, and a group of Italian patients with ACM to understand how different DSG2 changes affect disease risk. We found that disease severity depends not only on the type of genetic change but also on the location within the gene and whether other related genes are involved. These findings improve understanding of inherited heart disease and support more accurate genetic testing and follow-up for affected families.
Similar content being viewed by others

Data availability
The full-length DSG2 protein structure was obtained from the AlphaFold Protein Structure Database (UniProt accession: Q14126; model AF-Q14126-F1, version v4). DSG2 genetic variants were systematically collected from publicly available databases, including gnomAD v4.1.0 (https://gnomad.broadinstitute.org) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). No newly generated sequencing data were deposited in public repositories. Clinical data, including ECG, echocardiography, and CMR evaluations, were collected according to the current ESC 2023 Guidelines. Due to ethical and privacy constraints related to human genetic and clinical data, the raw data generated and analyzed in this study are not publicly available. Access to the raw data may be granted upon reasonable request and subject to approval by the corresponding author, in accordance with institutional policies and applicable regulations. Requests should be addressed to Kalliopi Pilichou (email: kalliopi.pilichou@unipd.it). A response to data access requests will be provided within 15 working days of receipt. Approved access will be subject to a data use agreement defining conditions of use, including restrictions on data sharing, requirements for appropriate data protection, and limitation of use to non-commercial research purposes.
References
Green, K. J. & Simpson, C. L. Desmosomes: new perspectives on a classic. J. invest. Dermatol. 127, 2499–2515 (2007).
Sikora, M. et al. Desmosome architecture derived from molecular dynamics simulations and cryo-electron tomography. Proc. Natl. Acad. Sci. USA 117, 27132–27140 (2020).
Delva, E., Tucker, D. K. & Kowalczyk, A. P. The desmosome. Cold Spring Harb. Perspect. Biol. 1, a002543 (2009).
Schafer, S., Stumpp, S. & Franke, W. W. Immunological identification and characterization of the desmosomal cadherin Dsg2 in coupled and uncoupled epithelial cells and in human tissues. Differentiation 60, 99–108 (1996).
Schafer, S., Koch, P. J. & Franke, W. W. Identification of the ubiquitous human desmoglein, Dsg2, and the expression catalogue of the desmoglein subfamily of desmosomal cadherins. Exp. Cell Res. 211, 391–399 (1994).
Schlipp, A. et al. Desmoglein-2 interaction is crucial for cardiomyocyte cohesion and function. Cardiovasc. Res. 104, 245–257 (2014).
Carvalho, S., Reis, C. A. & Pinho, S. S. Cadherins glycans in cancer: sweet players in a bitter process. Trends Cancer 2, 519–531 (2016).
Kolegraff, K., Nava, P., Laur, O., Parkos, C. A. & Nusrat, A. Characterization of full-length and proteolytic cleavage fragments of desmoglein-2 in native human colon and colonic epithelial cell lines. Cell Adh Migr. 5, 306–314 (2011).
Cason, M. et al. Novel pathogenic role for galectin-3 in early disease stages of arrhythmogenic cardiomyopathy. Heart Rhythm 18, 1394–1403 (2021).
Gupta, A. et al. Cell cycle- and cancer-associated gene networks activated by Dsg2: evidence of cystatin A deregulation and a potential role in cell-cell adhesion. PLoS One 10, e0120091 (2015).
Myo Min, K. K. et al. Desmoglein-2 as a cancer modulator: Friend or foe? Front. Oncol. 13, 1327478 (2023).
Pilichou, K. et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113, 1171–1179 (2006).
James, C. A. et al. An international evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy (ARVC) using the ClinGen framework. Circ. Genom. Precis. Med. 14, e003273 (2021).
Marcus, F. I. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 121, 1533–1541 (2010).
Corrado, D. et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int. J. Cardiol. 319, 106–114 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Hershberger, R. E. et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 20, 899–909 (2018).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. Off. J. Am. Coll. Med. Genet. 22, 245–257 (2020).
Pejaver, V. et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 109, 2163–2177 (2022).
Stenton, S. L. et al. Assessment of the evidence yield for the calibrated PP3/BP4 computational recommendations. Genet. Med. Off. J. Am. Coll. Med. Genet. 26, 101213 (2024).
Liu, S., Feng, X., Wu, Y. & Bu, F. Calibration and refinement of ACMG/AMP criteria for variant classification with BayesQuantify. J. Med. Genet. https://doi.org/10.1136/jmg-2025-110863 (2025).
Arbelo, E. et al. 2023 ESC guidelines for the management of cardiomyopathies. Eur. Heart J. 44, 3503–3626 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Hu, X., Dong, Q., Yang, J. & Zhang, Y. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 32, 3694 (2016).
Rodrigues, C. H., Pires, D. E. & Ascher, D. B. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46, W350–W355 (2018).
Delport, W., Poon, A. F., Frost, S. D. & Kosakovsky Pond, S. L. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457 (2010).
Rasmussen, T. B. et al. Mutated desmoglein-2 proteins are incorporated into desmosomes and exhibit dominant-negative effects in arrhythmogenic right ventricular cardiomyopathy. Hum. Mutat. 34, 697–705 (2013).
Gaertner, A. et al. Myocardial transcriptome analysis of human arrhythmogenic right ventricular cardiomyopathy. Physiol. Genomics 44, 99–109 (2012).
Kant, S., Holthofer, B., Magin, T. M., Krusche, C. A. & Leube, R. E. Desmoglein 2-dependent arrhythmogenic cardiomyopathy is caused by a loss of adhesive function. Circ. Cardiovasc. Genet. 8, 553–563 (2015).
Qadri, S. et al. Case reports of two pedigrees with recessive arrhythmogenic right ventricular cardiomyopathy associated with homozygous Thr335Ala variant in DSG2. BMC Med. Genet. 18, 86 (2017).
Quarta, G. et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 123, 2701–2709 (2011).
Bidina, L. et al. PKP2 and DSG2 genetic variations in Latvian arrhythmogenic right ventricular dysplasia/cardiomyopathy registry patients. Anatol. J. Cardiol. 20, 296–302 (2018).
Chen, L. et al. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int. J. Cardiol. 274, 263–270 (2019).
Mendell, J. T. & Dietz, H. C. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell 107, 411–414 (2001).
Eshkind, L. et al. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol. 81, 592–598 (2002).
Rigato, I. et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 6, 533–542 (2013).
Yesudian, P. D. et al. Novel compound heterozygous mutations in the desmoplakin gene cause hair shaft abnormalities and culminate in lethal cardiomyopathy. Clin. Exp. Dermatol 39, 506–508 (2014).
Nakajima, T. et al. Compound and digenic heterozygosity in desmosome genes as a cause of arrhythmogenic right ventricular cardiomyopathy in Japanese patients. Circ. J. 76, 737–743 (2012).
Jain, A., Sharma, D., Bajaj, A., Gupta, V. & Scaria, V. Founder variants and population genomes-Toward precision medicine. Adv. Genet. 107, 121–152 (2021).
Bao, J. R. et al. Screening of pathogenic genes in Chinese patients with arrhythmogenic right ventricular cardiomyopathy. Chin. Med. J. 126, 4238–4241 (2013).
Wada, Y., Ohno, S., Aiba, T. & Horie, M. Unique genetic background and outcome of non-Caucasian Japanese probands with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Mol. Genet. Genom. Med. 5, 639–651 (2017).
Murakami, H. et al. Arrhythmogenic right ventricular cardiomyopathy in a Japanese patient with a homozygous founder variant of DSG2 in the East Asian population. Hum. Genome Var. 9, 28 (2022).
van Lint, F. H. M. et al. Arrhythmogenic right ventricular cardiomyopathy-associated desmosomal variants are rarely de novo. Circ. Genom. Precis. Med. 12, e002467 (2019).
Pilichou, K. et al. Large genomic rearrangements of desmosomal genes in italian arrhythmogenic cardiomyopathy patients. Circ Arrhythm Electrophysiol 10, e005324 (2017).
Brodehl, A. et al. Hemi- and homozygous loss-of-function mutations in DSG2 (Desmoglein-2) cause recessive arrhythmogenic cardiomyopathy with an early onset. Int. J. Mol. Sci. 22, 3786 (2021).
Xu, T. et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 55, 587–597 (2010).
Vite, A. et al. Desmosomal cadherins are decreased in explanted arrhythmogenic right ventricular dysplasia/cardiomyopathy patient hearts. PLoS One 8, e75082 (2013).
Zou, Y. et al. A common indel polymorphism of the Desmoglein-2 (DSG2) is associated with sudden cardiac death in Chinese populations. Forensic Sci. Int. 301, 382–387 (2019).
Fressart, V. et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace: European pacing, arrhythmias, and cardiac electrophysiology: journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology. Eur. Soc. Cardiol. 12, 861–868 (2010).
Bhuiyan, Z. A. et al. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: results from a multicenter study. Circ. Cardiovasc. Genet. 2, 418–427 (2009).
Chen, L. et al. Natural history and clinical outcomes of patients with DSG2/DSC2 variant-related arrhythmogenic right ventricular cardiomyopathy. Circulation 151, 1213–1230 (2025).
Hermida, A. et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Heart Fail 21, 792–800 (2019).
Jorda, P. et al. Arrhythmic risk prediction in arrhythmogenic right ventricular cardiomyopathy: external validation of the arrhythmogenic right ventricular cardiomyopathy risk calculator. Eur. Heart J. 43, 3041–3052 (2022).
Jordan, E. et al. An evidence-based assessment of genes in dilated cardiomyopathy. Circulation 144, 7–19 (2021).
Ng, K. E. et al. Early inflammation precedes cardiac fibrosis and heart failure in desmoglein 2 murine model of arrhythmogenic cardiomyopathy. Cell Tissue Res. 386, 79–98 (2021).
Chelko, S. P. et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 140, 1491–1505 (2019).
Ammirati, E. et al. Acute myocarditis associated with desmosomal gene variants. JACC Heart Fail 10, 714–727 (2022).
Bhonsale, A. et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 36, 847–855 (2015).
Murray, B. & James, C. A. Genotype-phenotype correlates in arrhythmogenic cardiomyopathies. Curr. Cardiol. Rep. 24, 1557–1565 (2022).
Hawthorne, R. N. et al. Altered electrical, biomolecular, and immunologic phenotypes in a novel patient-derived stem cell model of desmoglein-2 mutant ARVC. J. Clin. Med. 10, 3061 (2021).
Noor Ul Ayan, H. et al. Homozygous frameshift variant in desmoglein 2 gene causes biventricular arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 104, 266–268 (2023).
Kapplinger, J. D. et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J. Am. Coll. Cardiol. 57, 2317–2327 (2011).
Lin, Y. et al. Whole genome sequence identified a rare homozygous pathogenic mutation of the DSG2 gene in a familial arrhythmogenic cardiomyopathy involving both ventricles. Cardiology 138, 41–54 (2017).
Awad, M. M. et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 79, 136–142 (2006).
Bauce, B. et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 7, 22–29 (2010).
Chen, P. et al. Distal myopathy induced arrhythmogenic right ventricular cardiomyopathy in a pedigree carrying novel DSG2 null variant. Int. J. Cardiol. 298, 25–31 (2020).
Dalal, D. et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation 113, 1641–1649 (2006).
Gaertner, A. et al. In vitro functional analyses of arrhythmogenic right ventricular cardiomyopathy-associated desmoglein-2-missense variations. PLoS One 7, e47097 (2012).
Harrison, O. J. et al. Structural basis of adhesive binding by desmocollins and desmogleins. Proc. Natl. Acad. Sci. USA 113, 7160–7165 (2016).
Debus, J. D. et al. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell Cardiol. 129, 303–313 (2019).
Gehmlich, K. et al. Novel missense mutations in exon 15 of desmoglein-2: Role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm 7, 1446–1453 (2010).
Chen, J. et al. The C-terminal unique region of desmoglein 2 inhibits its internalization via tail-tail interactions. J. Cell Biol. 199, 699–711 (2012).
Acknowledgements
This work was supported by the Italian Ministry of University and Research (MUR) (PRIN grant 20229FE439—CUP C53D23004670006 “The mechanistic link between genetic substrate and immune reactions in inflammatory cardiomyopathies”), Rome; the Registry for Cardio-cerebro-vascular Pathology, Veneto region, Venice; and ARCA (Associazione Ricerche Cardiopatie Aritmiche), Padova, Italy. RC and MBM are Postdoc Fellows supported by the Italian Ministry of Health, PNRR Next-Generation EU grant PNRR-MR1-2022-12376614—CUP I93C22000570006 “Machine learning approach for inherited Arrhythmic cardiomyopathies re-classification and risk stratification: from imaging to genomics. “ Funded by the European Union—Next Generation EU—NRRP M6C2—Investment 2.1 “Enhancement and strengthening of biomedical research in the NHS.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.P. Formal analysis: S.P., M.C., M.B.M., and R.C. Funding acquisition: K.P., C.B., D.C., and G.Th. Investigation: S.P. Methodology: S.P., M.C., M.B.M., R.C., F.D.Z., G.T., and M.D.G. Clinical investigation: M.M., I.R., S.R., M.D.G., B.B., and C.B. Resources: K.P. and C.B. Software: S.P., M.C., M.B.M., and R.C. Supervision: K.P. Writing original draft: S.P. and K.P. Writing—review and editing: B.B., G.Th., C.B., and K.P. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Roddy Walsh, Jasper J. van der Smagt, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pinci, S., Celeghin, R., Martini, M. et al. Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation. Commun Med (2026). https://doi.org/10.1038/s43856-026-01416-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43856-026-01416-w
