
Abstract
Limited cycle life remains a major obstacle to the practical application of high-capacity alloy negative electrodes in rechargeable batteries aimed at boosting energy density. The key challenges lie in the inherent uncontrollable volume changes and unstable electrode-electrolyte interphases. Here, we demonstrate a long-life self-construal tin (Sn) negative electrode for sodium (Na)-ion batteries enabled by in situ-formed embedded C–N anchors that integrate mechanical and chemical restrictions. This effect reshapes the alloying reactions with pronounced phase transformation hysteresis and triggers an electrochemically driven self-reconstructed structure for alloying reactions, therefore resolving the uncontrollable volume expansion and associated detrimental effects. C–N anchors also promote the formation of unique viscoelastic electrode-electrolyte interphases that comfortably accommodate large volume fluctuations for thousands of cycles. The designed Sn-based negative electrode exhibits long cycle life over 7000 cycles with a low-capacity decay rate of ~0.0036% at 2 C. Stable cycling of the Sn-based negative electrode is further confirmed in a prototype Na-ion pouch cell. This work offers an efficient design of employing the inherent volume expansion of alloy to electrochemically induce a self-constructed structure that comfortably accommodates volume changes, thereby ensuring long cycle life.
Similar content being viewed by others


Introduction
Alloy-type negative electrodes hold great promise as negative electrode materials for both lithium (Li)-ion batteries and sodium (Na)-ion batteries because of their high capacity and low cost1,2. Compared with conventional insertion negative electrodes, alloys suffer critical challenges of the inherent large volume expansion during (de)alloying reactions, e.g., up to 420% for the alloying reaction of Sn with Na3,4. This issue causes particle fragmentation, loss of electrical contact, continuous rupture/reformation of electrode-electrolyte interphase layers, and inferior cycle life5,6,7. Uncontrollable volume expansion has fundamentally hindered the commercialization of alloy-type negative electrodes for decades8. Therefore, addressing this problem becomes paramount for promoting their potential in high-energy storage technologies.
Addressing the issues of volume expansion is subject to understanding the fundamental mechanisms and evolution of volume expansion. However, electrochemical/chemical-mechanical coupling challenges comprehensive understanding and rationalized design strategies. Various approaches, such as particle downsizing9,10, interface engineering11,12, conductive composites13,14,15, chemical modulation16,17, and electrolyte optimization18,19, have been proposed to mitigate the volume expansion. Despite improvements, most conventional approaches rely on nano-structuring, which often suffers from high surface area, increased manufacturing cost and low tap density20,21. Critically, these strategies lack a sustainable mechanism that can accommodate continuous volume variations for thousands of cycles while maintaining interfacial stability for practical applications.
We take Sn-based alloy negative electrodes for Na-ion batteries as a model in this work. Although Sn offers a high theoretical capacity (847 mAh g⁻¹) and natural abundance, its practical application is severely hindered by rapid capacity decay and structural pulverization during cycling. Unlike conventional nano-structuring or ex situ coating approaches, we propose a scalable in situ construction strategy to embed C–N anchors that provide collaborative chemical and mechanical confinements for the alloying reactions of Sn-based negative electrodes. Comprehensive in situ and ex situ characterizations reveal that these C–N anchors fundamentally reshape the alloying pathway and induce a self-adaptive evolution of the Sn electrode architecture. This adaptive mechanism effectively accommodates large volume changes, ensuring remarkable long-term cycling stability. Moreover, the C–N anchors promote the formation of a unique viscoelastic and chemically robust interphase, which sustains interfacial stability over thousands of cycles. This work offers fresh insights into the rational control of high-capacity alloying reactions for practical Na-ion batteries.
Results
Synthesis and characterizations of Sn/SnO2-CN
A precisely controlled thermal reduction strategy is developed to construct the interconnected metallic Sn embedded in the in situ formed C–N anchors derived from tyrosine, denoted as Sn/SnO2-CN (Fig. 1a; see Supplementary Note 1 for details). High surface tension of molten Sn drives the formation of micro-sized spherical particle morphology during synthesis (Supplementary Fig. 1)22. The synthesis process can be conveniently controlled via thermal reduction time, as evidenced by the X-ray diffraction (XRD) and scanning electron microscope (SEM) morphology characterizations in Fig. 1b and Supplementary Fig. 2. The optimized reduction time is about 3 h, which well balances in situ structural regulation and the active Sn content (Supplementary Fig. 3).

a Schematic illustration of synthesis procedures. b XRD patterns of Sn/SnO2-CN samples with different thermal reduction times. c 3D nano-tomography with synchrotron TXM of the Sn/SnO2-CN particles and cross-sectional displays for different depths. d TEM-EDS mapping of interior Sn/SnO2-CN sliced by FIB. e Corresponding content of each element and C–N anchor skeleton schematic diagram. f C 1 s and N 1 s peaks in the XPS survey. g HRTEM images for Sn/SnO2-CN slice obtained via FIB.
Nano-tomography with synchrotron transmission X-ray microscopy (TXM) reveals the internal structure within Sn/SnO2-CN (Fig. 1c). Homogeneous and dense Sn distribution can be verified through the depth profiles of the 2D cross-section. The lighter coloration at the surface zone is the result of residual unreacted nano SnO2 at the outer surface23 (Supplementary Fig. 4).
Energy-dispersive spectroscopy (EDS) imaging of the cross-section for Sn/SnO2-CN sliced by focused ion beam (FIB) exhibits a uniform distribution of Sn, N, C, and O at the nanoscale (Fig. 1d and Supplementary Fig. 5), indicating the simultaneous formation of percolating Sn phase embedded in C–N anchors. Elemental analysis indicates the molar ratios of 76.5% Sn, 14.0% N, 5.8% C and 3.7% O (Fig. 1e). The observation of C–C, C–N and various types of N species in X-ray photoelectron spectroscopy (XPS) spectra confirm the formation of C–N anchors derived from tyrosine carbonization during synthesis24,25 (Fig. 1f). Specifically, the presence of C–Sn and N–Sn bonds in the XPS spectra verifies strong chemical bonding interactions between Sn and C–N anchors. These chemical bonds effectively bridge the percolating Sn and C–N anchors which provide integrated chemical and mechanical regulations for the volume variation during (de)alloying with Na.
Interestingly, the in situ formed C–N anchors significantly modulate the crystallite Sn distribution within Sn/SnO2-CN at nanoscale, which exhibits a dense and alternating crystalline-disordered secondary structure extending from the surface to the core (Fig. 1g and Supplementary Fig. 6). This distinctive phase arrangement likely originates from the nanoconfinement effect imposed by the C–N framework, which spatially restricts the migration and coalescence of molten Sn during reduction, thereby preventing the formation of large crystalline aggregates. Compared with conventional microscale Sn particles, this unique alternating crystalline-disordered Sn could potentially enhance the damage tolerance against large volume changes during cycling.
Electrochemical performances of Sn/SnO2-CN in half-cells
Cyclic voltammetry (CV) measurement validates the (de)alloying reaction in Sn/SnO2-CN negative electrode, showing two prominent pairs of alloying redox peaks at 0.244 V/0.013 V and 0.550 V/0.126 V (Fig. 2a and Supplementary Fig. 7)26,27. The electrode delivers a high initial Coulombic efficiency (ICE) of approximately 82 ~ 85%, comparable to or exceeding that of many reported Sn-based negative electrodes (Supplementary Fig. 8 and Supplementary Table 1). Notably, Sn/SnO2-CN demonstrates high-rate rate capability with high reversible capacities of 750, 746, 722, 696, and 624 mAh g−1 at 0.2 C, 0.5 C, 1 C, 2 C, and 5 C, respectively (Fig. 2b). The embedded C–N anchors could be the main reason for the improved rate performance, which effectively maintains electrical contact and structural integrity despite large volume changes, even at high C-rates. Well-retained charge-discharge curves from 0.1 C to 5 C further confirm the favorable reaction kinetics of the Sn/SnO2-CN electrode (Fig. 2c). In contrast, the nano-Sn electrode exhibits rapid capacity decay at elevated rates, particularly at 5 C, which is mainly ascribed to severe polarization under high-rate operation (Fig. 2b and Supplementary Fig. 9). Meanwhile, the nano-SnO₂ electrode shows negligible redox activity compared with the metallic Sn-based electrodes (Fig. 2b and Supplementary Fig. 7).

a CV curves of Sn/SnO2-CN at the scan rate of 0.1 mV s−1. b Rate performances. c Galvanostatic discharge-charge curves of the Sn/SnO2-CN negative electrode at different C rates (1C = 0.847 A g−1). d Cycling performances of Sn-based electrodes at 2C. e Cycling performance of Sn/SnO2-CN at 5 C. f Galvanostatic discharge-charge curves of Sn/SnO2-CN at 5 C over 2000 cycles. g Comparison of the cycling performance of Sn/SnO2-CN with the reported typical negative electrode materials for Na-ion batteries17,28,29,30,31,32,33,34,35,36, the size of the circles indicates the magnitude of the C-rate during cycling. Detailed information has been provided in Supplementary Table 2.
In addition, Sn/SnO2-CN electrode delivers a prolonged lifespan of 7000 cycles at 2C, retaining 75% capacity and an average Coulombic efficiency of ~99.94% (Fig. 2d). Such long-term cycling stability is rarely achieved by conventional Sn-based negative electrodes in Na-ion batteries. On the other hand, the nano-Sn electrode initially delivers 595 mAh g−1 at 2 C but suffers from rapid capacity decay and cell failure after only 566 cycles, likely due to the loss of active materials and electric contact caused by uncontrolled volume expansion and structural pulverization (Supplementary Fig. 9). Even at higher rate of 5 C, Sn/SnO2-CN maintains a reversible capacity of ~660 mAh g−1, retaining over 85% of its capacity after 5000 cycles (Fig. 2e). The nearly identical charge-discharge curves over long-term cycling without significant overpotential increase further confirm the robust electrochemical stability of Sn/SnO2-CN (Fig. 2f). Cycling performances of Sn/SnO2-CN at various C rates and higher mass loading are presented in Supplementary Figs. 10, 11. Compared with the typical negative electrode materials reported in Na-ion batteries (Fig. 2g), Sn/SnO2-CN demonstrates a significant advantage in rate capability, capacity retention, and long-term cycling stability17,28,29,30,31,32,33,34,35,36.
Alloying reaction mechanism and 3D imaging of structural evolution
By combining in situ XRD and ex situ solid-state NMR (ss-NMR), alloying reaction mechanism in Sn/SnO2-CN is systematically investigated to elucidate the underlying structural stability. It is found that the embedded C–N anchors significantly influence the phase transition pathway of Sn/SnO2-CN during (de)alloying with Na. In situ XRD measurements show Sn/SnO2-CN undergoes similar initial phase transitions to nano-Sn electrode, progressively forming crystallite NaSn5 and an amorphous NaxSn phase (Fig. 3a, b and Supplementary Figs. 12, 13)37,38. 23Na ss-NMR spectra verifies the corresponding emergence of NaSn5 at ~50 ppm and amorphous NaxSn with a broad peak at ~72 ppm (Fig. 3c)39. As the reaction proceeds (point B-C), amorphous NaxSn gradually transforms into amorphous Na9Sn4 and subsequently into crystalline Na9Sn4, as evidenced by the emerging XRD reflections and a characteristic NMR signal at ~218 ppm (Fig. 3a–c)5,39. This phase evolution likely accounts for the small voltage bump observed during discharge in Fig. 2c at low C rate (see detailed discussion in Supplementary Note 2). Upon further alloying reaction with Na, Na9Sn4 continues to transform into the final alloying phase of Na15Sn4.

a In situ XRD spectra during the first cycle. b Voltage curve. c Ex situ 23Na ss-NMR spectra corresponding to points A–H at different SOC in (b). d–g 3D nano-tomography with synchrotron TXM for point C (d), point D (e), point G (f) and point H (g), and cross-sectional imaging at different depths. Scale bar: 2 μm. h-i X-ray attenuation ratio curves of three corresponding lines in Fig. e (h) and Fig. g (i). j Schematic diagram of the expansion procedures of Sn/SnO2-CN and nano-Sn. All the in/ex suit measurements were conducted during the first cycle at a specific current of 1.694 A g−1 (0.2C) and 25 °C.
Interestingly, the Na9Sn4 phase is nearly absent in the in situ XRD patterns at the end discharge (Fig. 3a), whereas the ex situ 23Na ss-NMR spectrum still displays a distinct Na9Sn4 resonance (~218 ppm) alongside the presence of Na15Sn4 (−225 and −343 ppm)40. These intriguing results indicate that the fully discharged Sn/SnO2-CN electrode does not completely convert to crystalline Na15Sn4 but instead retains a fraction of amorphous Na9Sn4 and Na15Sn4. This incomplete crystallization likely originates from the restricted atomic diffusion and inhibited long-range ordering imposed by the C–N anchoring network.
The delayed phase transition behavior is further supported by the de-alloying process. Upon initial charge (point D–E), the intensity of Na15Sn4 XRD peaks unexpectedly increases before diminishing, revealing a phase transition hysteresis induced by the C–N framework. The chemical anchoring and mechanical confinement of C–N anchors kinetically restrict the phase transition from Na9Sn4 to Na15Sn4, which essentially mitigates the volume expansion during discharge and contributes to the enhanced cycling stability. Sn/SnO2-CN undergoes a reversible phase transition, returning to metallic Sn during the subsequent charging process. While the residual SnO2 phase in Sn/SnO2-CN also presents a basically reversible change during charge and discharge. In contrast, in situ XRD measurement for nano-Sn exhibits a conventional phase transition process, with no observable phase transition hysteresis at the end of discharge (Supplementary Fig. 13).
Structural evolution of Sn/SnO2-CN at different alloying stages is further examined to understand how embedded C–N anchors accommodate large volume changes. Notes that Sn/SnO2-CN still exhibits minimal volume expansion until the later stages of alloying (~600 mAh g−1, Point C), while maintaining an intact particle morphology (Fig. 3d). Depth profiling reveals well-integrated internal structure, and the color difference may come from the local difference in the degree of alloying. At the fully discharged state (Point D), nano-tomography with synchrotron TXM for the Sn/SnO2-CN exhibits significant volume expansion but effectively retains the particle integrity without particle pulverization (Fig. 3e). Depth-profiling indicates the occurrence of a self-constructing 3D interconnected pore structure during alloying reactions, which is likely result of the mechanical confinement and chemical binding effects of the embedded C–N anchors within the Sn/SnO2-CN. During de-alloying (charging), the expanded particles reversibly contract, while the 3D porous network self-adjusts and recovers to an intact internal structure, ensuring reliable structural stability (Fig. 3f, g).
X-ray attenuation profiles further demonstrate the structural features of Sn/SnO2-CN at fully (de)alloyed states (Fig. 3h, i). Significant attenuation drops at internal pore regions, while lower attenuation values manifested as shoulder peaks and troughs are attributed to the existence of embedded C–N anchors. Notably, these attenuation signatures persist in both fully discharged and charged states, providing direct evidence for the stable existence of percolating C–N anchors within the material. The flexible C–N anchor skeleton demonstrates dynamic structural adjustments during the Sn phase transition and phase growth processes, which enable favorable volume expansion adaptability. Figure 3j elucidates the underlying mechanisms. Compared with conventional nano-Sn, Sn/SnO2-CN effectively dissipates volumetric expansion stresses through collective chemical anchoring and mechanical confinement effects exerted by the C–N anchors, thereby maintaining structural integrity during cycling.
A sustained structure capable of accommodating repeated large volume changes is crucial for long-term cycling stability of alloy negative electrodes. HRTEM images indicate that Sn/SnO2-CN still maintains intact particle morphology without fragmentation after cycling (Fig. 4a, b). A thin and uniform solid-electrolyte interphase (SEI) layer is formed on the particle surface of Sn/SnO2-CN in the first cycle and remains stable over extended cycles, which effectively prevents parasitic electrolyte reactions and helps preserve the internal conductive network for sustainable reversibility. The alternating distribution of crystalline-disordered features of Sn within Sn/SnO2-CN can be retained after repeated cycles (Supplementary Fig. 14), demonstrating strong structural reversibility. In contrast, the nano-Sn electrode exhibits disordered particle distributions, uneven and thick SEI layers, and severe particle cracking (Supplementary Fig. 15), which accounts for the continuous capacity degradation.

a, b HRTEM images of Sn/SnO2-CN negative electrodes after 1 cycle at 0.1C (a) and 100 cycles at 5C (b). c–f Ex situ 3D nano-tomography with synchrotron TXM and cross-sectional displays of pristine Sn/SnO2-CN (c), the negative electrodes after 1 cycle at 0.1 C (d), after 50 cycles at 5 C (e) and after 500 cycles at 5 C (f). Scale bar in Fig. 4c–f: 2 μm.
Structural durability and evolution of Sn/SnO2-CN are further analyzed over long cycles via synchrotron TXM (Fig. 4c–f). Color intensity in the images represents Sn distribution. The initial dense Sn/SnO2-CN evolves to an interconnected porous network after initial (de)alloy reactions as discussed in Fig. 4c, d and Supplementary video 1. With continued cycling, this pore structure gradually develops into a coral-like microsphere network with smaller and more uniform pores (Fig. 4e and Supplementary Fig. 16). This unique electrochemically induced self-reconstruction likely originates from the synergistic chemical anchoring and mechanical confinement effects of the embedded C–N network. These effects collectively regulate the phase transformation pathway and stress-release dynamics, effectively suppressing uncontrolled propagation of alloying products and imparting enhanced damage tolerance against volume fluctuations. As cycling proceeds, the coral-like microstructure becomes increasingly defined and compact, further enhancing mechanical robustness and structural stability (Fig. 4f and Supplementary Video 2). SEM and EDS-mapping analyses corroborate the TXM observations, confirming the progressive structural reconstruction and robust electrode-level stability of Sn/SnO2-CN during prolonged cycling (Supplementary Figs.17–19). This self-constructed architecture not only accommodates volume expansion but also enhances Na⁺ transport and electronic conductivity, improving rate capability and ensuring long-term cycling stability over thousands of cycles.
Interfacial chemical and mechanical stability of Sn/SnO2-CN
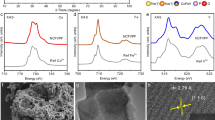
Intensive volume expansion can cause repeated cracking or delamination of the SEI layer, exposing fresh active surfaces, leading to continuous electrolyte consumption and cell performance degradation12,41. The interfacial chemistry and mechanical properties and their tolerance to repeated volume change are further studied for Sn/SnO2-CN. Depth-profiling XPS is employed to analyze SEI composition and formation (Fig. 5a–c, Supplementary Fig. 20). The SEI layer primarily consists of organic species (C-OR/C–N, C–F) and inorganic NaF. The organic C/O/F species likely originate from electrolyte decomposition or polymerization on the electrode surface12, while NaF results from Na salt decomposition17,42. After etching 20 nm, the organic carbon content significantly decreases, while NaF content increases, enhancing mechanical strength and ionic conductivity of the SEI layer43,44. Organic C-OR/C–N components spread throughout the SEI layer, ensuring necessary mechanical flexibility. Notably, the SEI layer of Sn/SnO2-CN also contains abundant N-containing species (Fig. 5c), with pyrrolic-N and pyridinic-N dominating the outer layer, while graphitic-N and N–Sn bonds become prominent in the inner layer.

a–c Depth profiling XPS spectra for C 1 s (a), F 1s (b) and N 1 s (c) of Sn/SnO2-CN electrode after 1 cycle at 0.1 C and 100 cycles at 5 C. d-e DRT curves of Sn/SnO2-CN (d) and nano-Sn (e) during long-term cycling. f 2D mapping of Young’s modulus derived from AFM measurements. g–h Typical F–D curves of Sn/SnO2-CN (g) and nano-Sn (h). i Schematic illustration of SEI during expansion and contraction process.
Since the electrolyte does not contain N, the N species in the SEI layer indicate that the C–N anchors in the Sn/SnO2-CN play a critical role in SEI formation. These N species act as chemical anchors, tightly integrating the SEI layer with Sn/SnO2-CN and preventing delamination or cracking during repeated volume changes. Even after 100 and 1000 cycles, the SEI layer retains its structure and composition (Supplementary Figs. 21, 22). XPS analysis confirms that the N-enriched SEI, featuring an organic-rich outer layer and an inorganic F-rich inner layer, remains robust and stable throughout long-term cycling. The durable SEI layer prevents repeated interface reconstruction and electrolyte decomposition, aligning with the stable electrochemical performance of Sn/SnO2-CN. In contrast, the nano-Sn electrode, lacking effective chemical anchoring and structural confinement during SEI formation, exhibits pronounced compositional disparities between the inner and outer layers, indicative of recurrent SEI rupture and regeneration (Supplementary Fig. 23), thereby accelerating performance degradation.
Dynamic interfacial changes of different Sn-based electrodes are studied using distribution of relaxation time (DRT) analysis over extended cycles. Peaks at different time constants in the DRT spectra indicate various interfacial processes45. The interfacial processes of the Sn/SnO2-CN electrode primarily include SEI layer resistance (RSEI), charge transfer resistance (Rct), and diffusion resistance (Rdiffu), as shown in Fig. 5d and Supplementary Fig. 24a. Consistently, the Sn/SnO2-CN electrode maintains low RSEI and Rct throughout long-term cycling with nearly unchanged DRT curves over 500 cycles, confirming the robust stability of SEI layer. On the other hand, the interfacial impedance of nano-Sn exhibits continuous fluctuations and an increasing trend during cycling, indicating poor SEI layer stability (Fig. 5e and Supplementary Fig. 24b).
Deformation resistance of the SEI layer is crucial for maintaining electrode integrity for alloy-based negative electrode. Using atomic force microscopy (AFM), the mechanical properties of the SEI layer on the electrode surface are studied under different states (Fig. 5f, Supplementary Fig. 25). The average Young’s modulus of SEI layer is approximately 2862 MPa at the first discharge capacity to 600 mAh g−1. Upon full discharge, significant volume expansion flattens the SEI layer and shows an increased value of 3634 MPa. The increased Young’s modulus at the end of discharge may result from the aligning order of stretched polyether chain segments in the SEI layer on the Sn/SnO2-CN surface46, which enhances intermolecular interactions, thereby improving Young’s modulus of the SEI layer. Moreover, the abundance of inorganic NaF and N-bonding in the SEI layer provides enhanced mechanical robustness for long cycle life17. Through repeated charge-discharge cycles, Sn/SnO2-CN develops an adaptive electrode structure and surface chemistry, underpinning its durable electrochemical stability. Conversely, nano-Sn exhibits significantly lower and unevenly distributed modulus values (Supplementary Fig. 26), likely due to repeated SEI rupture, reformation, and poor coverage.
Further comparison of force-deformation (F-D) curves reveals that Sn/SnO2-CN exhibits distinct strain hysteresis and viscoelastic deformation34,47, forming a characteristic viscoelastic loop in its loading-unloading curves (Fig. 5g). This behavior likely arises from the sliding/stretching of polyether chains in the SEI layer48,49, originating from the chemical anchoring effects of N species and NaF. The enclosed area of the viscoelastic loop corresponds to the energy dissipated during deformation. Such viscoelasticity enables the SEI layer to deform adaptively rather than fracture under large volume fluctuations50, thereby preserving interfacial integrity and enabling enhanced damage tolerance and cycling stability, as discussed above. In contrast, nano-Sn exhibits steep, nearly overlapping loading-unloading curves (Fig. 5h, Supplementary Fig. 27), suggesting the SEI layer formed on nano-Sn is rigid with limited deformation resistance47. Figure 5i illustrates the functions of the viscoelastic SEI layer on the Sn/SnO2-CN electrode during the (de)alloying processes. The unique structure and C–N anchors of Sn/SnO2-CN promote the formation of a robust and adaptive SEI layer that comfortably accommodates volume changes, enabling stable electrochemical performance in Sn-based alloy negative electrodes.
Validation of Sn/SnO2-CN negative electrode in full cells
We furthermore evaluate the electrochemical performance of Sn/SnO2-CN in full-cell configurations using Na3V2(PO4)3/C (NVP/C) as the positive electrode (Fig. 6a). Sn/SnO2-CN-based full cell achieves a high initial reversible capacity of 678 mAh g−1 at 0.2 C (Supplementary Fig. 28). It also exhibits favorable rate performance, maintaining reversible capacities of 484, 454, and 423 mAh g−1 at 2C, 5C, and 10C, respectively, with full capacity recovery after high-rate tests (Fig. 6b). In contrast, the nano-Sn full cell delivers only 497 mAh g−1 at 0.2C and suffers from rapid capacity decay at higher rates. The capacity of nano-Sn electrode fails to recover upon returning to lower rates due to poor structural stability. A Ragone plot comparison (Supplementary Fig. 29) highlights the pronounced advantages in energy and power densities of the Sn/SnO2-CN-based full cell. Additionally, Sn/SnO2-CN full cell demonstrates an initial capacity of ~489 mAh g−1 at 5 C, with negligible capacity loss over 500 cycles (Fig. 6c). Even after 2000 cycles, the full cell remains a high-capacity retention of 78%. In contrast, the nano-Sn full cell starts with a lower initial capacity of 173 mAh g−1 at 5 C, which rapidly declines to 129 mAh g−1 within just 100 cycles.

a Schematic configuration of Na-ion full cell with Sn/SnO2-CN negative electrode and NVP/C positive electrode. b Rate performances in coin-type Sn/SnO2-CN||NVP/C and nano-Sn||NVP/C full cells. c Cycle performance of typical coin-type full cells at 5 C. d Cycling performance of Sn/SnO2-CN||NVP/C pouch cell with corresponding charge/discharge curves at 2 C and the sizes of single-layer pouch cell. The full cell capacity is calculated based on the mass of negative electrode.
In addition, a prototype single-layer pouch cell with Sn/SnO2-CN as the negative electrode and NVP/C as the positive electrode is demonstrated in Fig. 6d. Without presodiation, the pouch cell delivers a high reversible capacity of 585 mAh g−1 at 0.2C and an average operating voltage of ~3.1 V in the first cycle (Supplementary Fig. 30), demonstrating pronounced performance. At 2C, the pouch cell maintains a reversible capacity of 449 mAh g−1 with reliable cycling stability over 400 cycles with negligible capacity degradation. The highly consistent charge-discharge profiles highlight the effectiveness of the C–N anchor-induced self-constructed structure in achieving long cycle life and high capacity for Sn-based alloy negative electrodes in Na-ion batteries.
Discussion
In this study, we report a long-life self-construal Sn negative electrode enabled by an in situ formed C–N anchor from a thermal reduction method for long-life and high-energy Na-ion batteries. The C–N anchors naturally integrate mechanical and chemical restrictions for Sn negative electrode, which can be refined by thermal reduction conditions. Combining in situ and ex situ observations, we reveal the critical role of integrated restrictions in reshaping alloying reactions with pronounced phase transition hysteresis during (de)alloying with Na and triggering electrochemically driven self-reconstructed structure that addresses uncontrollable volume expansion and enhances structural durability. Synchrotron X-ray 3D TXM unravels a sustainably self-adaptive volume change within the designed Sn negative electrode. In addition, the C–N anchors meanwhile promote the formation of unique viscoelastic electrode-electrolyte interfacial layers to accommodate the large volume change for thousands of cycles. The designed Sn alloy negative electrode demonstrates a high capacity of 750 mAh g−1 at 0.2C and long-term cycling stability over 7000 cycles at 2C with an average Coulombic efficiency of ~99.94%. Paired with NVP/C positive electrode, stable cycling is also validated in a prototype pouch cell. These findings provide insight into adaptable self-construal for alloying reactions by introducing integrated mechanical and chemical restrictions for developing long-life alloy negative electrodes.
Methods
Synthesis of Sn/SnO2-CN
First, spherical nano-sized SnO2 (Energy Chemical, 99.9% metals basis) and L-tyrosine (Maclin, 99%) were dispersed in deionized water with a mass ratio of 10:3 and stirred at 30 °C for 16 h to obtain Tyr@SnO2. Then, Tyr@SnO2 was ground with a mortar and annealed at 600 °C for 1–5 h in a 5% H2/Ar atmosphere to obtain Sn/SnO2-CN with a heating rate of 2 °C min−1, followed by natural cooling. A thermal reduction duration of 3 h was found to be optimal for a batch size of 0.2–1 g. It should be noted that process scaling may require further optimization, as larger batch sizes could alter heat and mass transfer behavior, thereby affecting the uniformity of reduction reactions and the formation of C–N anchors.
Synthesis of Na3V2(PO4)3/C (NVP/C) Composite
The synthesis was referred to the literature report51. In brief, vanadium pentoxide, anhydrous oxalic acid, glucose, sodium oxalate, and ammonium dihydrogen phosphate were dissolved in water at 80 °C and then dried slowly at 100 °C. After the powder had dried completely, the powder was further ground and annealed at 750 °C for 8 h in an Ar atmosphere.
Comparison samples
Nano-sized Sn powder (CANSPEC China, ≤100 nm, 99.9% metals basis) and SnO2 (Energy Chemical, <100 nm, 99.9% metals basis) were directly used without any purification as the control sample.
Material Characterizations
Ex situ and in situ XRD were characterized on an X-ray diffractometer (Rigaku, Ultima IV, Cu Kα radiation) with a scan rate of 5° min−1 and a step size of 0.02°. SEM images were collected on HITACHI SU8010 and beam voltage was set to 15 kV for EDX test. TEM images were collected on JEM2100F operating at 200 kV. XPS measurements were carried out with a K-Alpha (Thermo Scientific Al Kα, 1486.6 eV). The binding energies were calibrated by referencing the adventitious C 1s peak to 284.8 eV. For XPS measurements, the cycled cells were disassembled in an argon-filled glove box. The harvested electrodes were rinsed with DEGDME solvent to remove residual electrolytes and dried under vacuum. Since the XPS chamber was connected to the glove box, the sample transfer was performed without exposure to air. Similar disassembly and cleaning procedures were applied for other ex situ measurements. Cross-sections of Sn/SnO2-CN samples were obtained using a FIB/SEM (Quata 3D, FEG, Ga liquid metal). The elemental distribution on the cross-section was performed using EDS mapping. The internal microstructure and chemical composition of the Sn/SnO2-CN sample were further characterized for an FIB thinned Sn/SnO2-CN sample (~50nm) using HRTEM and TEM/EDS mapping (Tecnai G2 F20 S-TWIN).
Electrochemical measurements
Electrochemical performances of Sn-based negative electrodes were evaluated using 2032-type coin cells and single-layer pouch cells. The employed electrolyte was 1.0 M NaPF6 (battery grade, Hairong Power Material) in diethylene glycol dimethyl ether (battery grade, DoDoChem). The solvent was dried with 4 Å molecular sieves for 3 days in an argon-filled glove box to remove trace water before use. The Na metal counter electrodes used in half-cell tests were prepared by rolling and pressing fresh bulk Na metal into a flat and smooth plate (Thickness: ~200 μm). Then punch into round disks with a diameter of 15 mm before use. The preparation of Na electrode, the dissolution of electrolyte, and the assembly of cells were all carried out in an argon-filled glove box. For coin-type cells, the active material, conductive carbon (Super P) and binder (negative electrode: CMC, positive electrode: PVDF) with a specific weight ratio of (7:2:1 for half-cell, 8:1:1 for full-cell) was thoroughly mixed to form a uniform slurry, which was then tape cast onto Cu (~12 μm)/Al (~15 μm) foils and vacuum dried to obtain the single-side coated electrodes. Then punch into round disks with a diameter of 12 mm using the MSK-T10 manual slicer. The average mass loading of Sn-based materials was approximately 1.5 mg cm−2 (~30 μm), and NVP/C positive electrode mass loading was approximately 12 mg cm−2 (~150 μm) unless otherwise specified. Glass fiber separator (GF/F for half cells, 390 μm; GF/D for full cells, 540 μm) was punched into round disks with a diameter of 19 mm for use. CR2032 coin cells were assembled using stainless steel cases and springs (spring constant not calibrated). The electrolyte usages per coin- cell was 120 μL. At least three coin-cells were tested for each experimental condition to ensure reproducibility. A rate test from 0.2C to 5C was conducted for the full cell testing before cycling at 5C. The Coulombic efficiency (CE) was calculated based on the specific cell configuration: for half cells, CE is defined as the ratio of charge capacity to discharge capacity; for full cells, CE is defined as the ratio of discharge capacity to charge capacity.
For the single-layer pouch-type cell, Sn/SnO2-CN electrode (56 × 43 mm) was paired with NVP/C electrode (58 × 45 mm) at a negative to positive capacity (N/P) ratio of ~1.05. Glass fiber separator (Olegeeino, GF/A2916, thickness: 0.62 μm) was cut into 60*50 mm before use. The electrolyte usage was 1 g. Electrolyte injection was performed in an Ar-filled glove box, followed by primary vacuum sealing. The pouch cells were sandwiched between two 8 × 120 × 160 mm aluminum alloy plates held by four M6 bolts (Canrd, CF-TES-SPR08120160A) with applied external pressure of ~100 kPa during cycling. The electrochemical performances were tested on a NEWARE battery test system at a constant temperature of 25 °C in voltage ranges of 0.01–1.2 V for half cells and 2–3.8 V for full cells at different C rates (1C = 0.847 A g−1).
EIS measurements were performed on an electrochemical workstation (VSP300, Biologic, France) in potentiostatic mode with a frequency range from 1 MHz to 100 mHz, an amplitude of 5 mV and a data collection density of 6 points per decade of frequency. CV measurement was acquired in a voltage range from 0 V to 1.2 V at a scan rate of 0.1–1.2 mV s−1. Distribution of relaxation times (DRT) analysis was performed using the open-source DRT-TOOLS package developed by Ciucci et al., running within the MATLAB R2023a environment. This toolbox is publicly accessible under the GNU license at https://sites.google.com/site/drttools/.
Nano-tomography with synchrotron TXM
3D nano-tomography data based on synchrotron X-ray were collected at beamline BL18B of Shanghai Synchrotron Radiation Facility (SSRF)52,53. Each sample was characterized by 1021 projection images spanning an angular range from -90° to 90° at 8500 eV, providing a high spatial resolution of approximately 20 nm. The 3D nano-tomography reconstructions were generated from these 2D images using in-house code running on Python. TXM was employed to visualize the C–N network and pore distribution based on differences in X-ray attenuation coefficients. Sn-containing regions exhibit strong attenuation due to their high atomic number and density, whereas the C–N framework and pores display weak or near-zero attenuation. This contrast was used to correlate the morphology and structure of Sn/SnO2-CN. In attenuation line profiles, sharp valleys correspond to pores, while broader, moderately low-attenuation regions can be attributed to the C–N framework.
Characterizations and analysis
Topography of the cycled electrodes was obtained with atomic force microscopy (AFM, Dimension Icon, BRUKER) in the peak force tapping mode with sharp AFM tips (Bruker ScanAsyst Air) in an Ar-filled glove box. The modulus distribution of the SEI layer above the electrodes was obtained in peak force quantitative nanomechanical mapping (QNM) mode with sharp AFM tips (Bruker RTESPA-300). For the peak force QNM mode, the indentation depth was set at 2 nm.
Magic angle spinning (MAS) solid-state nuclear magnetic resonance (NMR) was conducted on a Bruker Avance III HD spectrometer operating at 400 MHz. A 3.2 mm probe was employed with a magic-angle spinning (MAS) rate of 15 kHz. Data acquisition utilized a Hahn-echo pulse sequence (90°-τ-180°-τ-acq) with a delay time (τ) of 64 μs. The 23Na spectra are referenced to 1 M NaCl solution at 0 ppm. The line shape deconvolution of SSNMR patterns was performed with the fitting module in Topspin.
Data availability
The data supporting the findings of this work are available within the article and its Supplementary Information files. Source data are provided with this paper.
References
Lu, Z. et al. Consummating ion desolvation in hard carbon anodes for reversible sodium storage. Nat. Commun. 15, 3497 (2024).
Wan, Y. et al. Fast-charging anode materials for sodium-ion batteries. Adv. Mater. 36, e2404574 (2024).
Lao, M. et al. Alloy-based anode materials toward advanced sodium-ion batteries. Adv. Mater. 29 (2017).
Chayambuka, K., Mulder, G., Danilov, D. L. & Notten, P. H. L. Sodium-ion battery materials and electrochemical properties reviewed. Adv. Energy Mater. 8 (2018).
Palaniselvam, T. et al. Sodium storage and electrode dynamics of tin–carbon composite electrodes from bulk precursors for sodium-ion batteries. Adv. Funct. Mater. 29 (2019).
Wang, J. W., Liu, X. H., Mao, S. X. & Huang, J. Y. Microstructural evolution of tin nanoparticles during in-situ sodium insertion and extraction. Nano Lett. 12, 5897–5902 (2012).
Tian, Y., An, Y. & Zhang, B. Approaching microsized alloy anodes via solid electrolyte interphase design for advanced rechargeable batteries. Adv. Energy Mater. 13 (2023).
Chen, J. et al. Electrolyte design for LiF-rich solid–electrolyte interfaces to enable high-performance microsized alloy anodes for batteries. Nat. Energy 5, 386–397 (2020).
Wang, X.-L., Feygenson, M., Aronson, M. C. & Han, W.-Q. Sn/SnOx core−shell nanospheres: synthesis, anode performance in Li ion batteries, and superconductivity. J. Phys. Chem. C 114, 14697–14703 (2010).
Liu, Y., Zhang, N., Jiao, L. & Chen, J. Tin nanodots encapsulated in porous nitrogen-doped carbon nanofibers as a free-standing anode for advanced sodium-ion batteries. Adv. Mater. 27, 6702–6707 (2015).
Liu, N. et al. A pomegranate-inspired nanoscale design for large-volume-change lithium battery anodes. Nat. Nanotechnol. 9, 187–192 (2014).
Huang, J. et al. Nanostructures of solid electrolyte interphases and their consequences for microsized Sn anodes in sodium ion batteries. Energy Environ. Sci. 12, 1550–1557 (2019).
Park, S.-H. et al. High areal capacity battery electrodes enabled by segregated nanotube networks. Nat. Energy 4, 560–567 (2019).
Ma, J. et al. Towards maximized volumetric capacity via pore-coordinated design for large-volume-change lithium-ion battery anodes. Nat. Commun. 10, 475 (2019).
Mo, R. et al. Tin-graphene tubes as anodes for lithium-ion batteries with high volumetric and gravimetric energy densities. Nat. Commun. 11, 1374 (2020).
Sung, J. et al. Subnano-sized silicon anode via crystal growth inhibition mechanism and its application in a prototype battery pack. Nat. Energy 6, 1164–1175 (2021).
Yang, J. et al. A high-performance alloy-based anode enabled by surface and interface engineering for wide-temperature sodium-ion batteries. Adv. Energy Mater. 13 (2023).
Zhang, B. et al. Microsized Sn as advanced anodes in glyme-based electrolyte for Na-ion batteries. Adv. Mater. 28, 9824–9830 (2016).
Kim, C. et al. A self-healing Sn anode with an ultra-long cycle life for sodium-ion batteries. J. Mater. Chem. A 6, 22809–22818 (2018).
Wu, C., Zhu, G., Wang, Q., Wu, M. & Zhang, H. Sn-based nanomaterials: from composition and structural design to their electrochemical performances for Li- and Na-ion batteries. Energy Storage Mater. 43, 430–462 (2021).
Qiao, S. et al. Advanced anode materials for rechargeable sodium-Ion Batteries. ACS Nano 17, 11220–11252 (2023).
Jain, S., John, A., George, C. E. & Johnson, R. P. Tyrosine-derived polymers as potential biomaterials: synthesis strategies, properties, and applications. Biomacromolecules 24, 531–565 (2023).
Wang, L., Wang, J. & Zuo, P. Probing battery electrochemistry with in-Operando synchrotron X-ray imaging techniques. Small Methods 2 (2018).
Jie, L. et al. The investigation of thermal decomposition pathways of phenylalanine and tyrosine by TG–FTIR. Thermochim. Acta 467, 20–29 (2008).
Osadchii, D. Y., Olivos-Suarez, A. I., Bavykina, A. V. & Gascon, J. Revisiting nitrogen species in covalent triazine frameworks. Langmuir 33, 14278–14285 (2017).
Sayed, S. Y., Kalisvaart, W. P., Luber, E. J., Olsen, B. C. & Buriak, J. M. Stabilizing tin anodes in sodium-ion batteries by alloying with silicon. ACS Appl Energy Mater. 3, 9950–9962 (2020).
Wu, X. et al. Tin-based anode materials for stable sodium storage: Progress and perspective. Adv. Mater. 34, e2106895 (2022).
Zhao, L. et al. Disodium terephthalate (Na2C8H4O4) as High performance anode material for low-cost room-temperature sodium-ion battery. Adv. Energy Mater. 2, 962–965 (2012).
Hu, Z. et al. MoS2 nanoflowers with expanded interlayers as high-performance anodes for sodium-ion batteries. Angew. Chem. 126, 13008–13012 (2014).
Wen, Y. et al. Expanded graphite as superior anode for sodium-ion batteries. Nat. Commun. 5 (2014).
Wu, L. et al. Unfolding the mechanism of sodium insertion in anatase TiO2 nanoparticles. Adv. Energy Mater. 5 (2014).
Cho, J. S., Park, J.-S. & Kang, Y. C. Porous FeS nanofibers with numerous nanovoids obtained by Kirkendall diffusion effect for use as anode materials for sodium-ion batteries. Nano Res 10, 897–907 (2016).
Wang, L. et al. Purifying the phase of NaTi2(PO4)3 for enhanced Na+ storage properties. ACS Appl. Mater. Interfaces 11, 10663–10671 (2019).
Lu, Z. et al. Step-by-step desolvation enables high-rate and ultra-stable sodium storage in hard carbon anodes. Proc. Natl. Acad. Sci. USA 119, e2210203119 (2022).
Yang, K. et al. Multi-yolk–shell Sn/Cu6Sn5@N–C nanospheres facilitate Na+/e– transfer at SEI, enabling 90.8% capacity retention at 10C. ACS Mater. Lett. 5, 2683–2690 (2023).
Yue, M. et al. Carbon-coated MOF-derived porous SnPS3 core-shell structure as superior anode for sodium-ion batteries. Small, e2405262 (2024).
Ellis, L. D., Hatchard, T. D. & Obrovac, M. N. Reversible insertion of sodium in tin. J. Electrochem. Soc. 159, A1801–A1805 (2012).
Ma, H. et al. Dynamic behavior of spatially confined Sn clusters and its application in highly efficient sodium storage with high initial Coulombic efficiency. Adv. Mater. 36, e2307151 (2024).
Stratford, J. M. et al. Investigating sodium storage mechanisms in tin anodes: a combined pair distribution function analysis, density functional theory, and solid-state NMR approach. J. Am. Chem. Soc. 139, 7273–7286 (2017).
Gotoh, K. 23Na solid-state NMR analyses for Na-ion batteries and materials. Batteries Supercaps 4, 1267–1278 (2021).
Tokranov, A. et al. Control and optimization of the electrochemical and mechanical properties of the solid electrolyte interphase on silicon electrodes in lithium ion batteries. Adv. Energy Mater. 6 (2016).
Yuan, H. et al. Ionic-conducting and robust multilayered solid electrolyte interphases for greatly improved rate and cycling capabilities of sodium-ion full cells. Adv. Energy Mater. 10 (2020).
Qin, B. et al. Highly reversible sodiation of tin in glyme electrolytes: The critical role of the solid electrolyte interphase and its formation mechanism. ACS Appl Mater. Interfaces 12, 3697–3708 (2020).
Zhang, X.-Q. et al. Regulating anions in the solvation sheath of lithium ions for stable lithium metal batteries. ACS Energy Lett. 4, 411–416 (2019).
Lu, Y., Zhao, C.-Z., Huang, J.-Q. & Zhang, Q. The timescale identification decoupling complicated kinetic processes in lithium-batteries. Joule 6, 1172–1198 (2022).
Tanzi MC, Farè S, Candiani G. Chapter 2 - Mechanical Properties of Materials. In: Foundations of Biomaterials Engineering (eds Tanzi MC, Farè S, Candiani G). (Academic Press, 2019).
Li, G. et al. Organosulfide-plasticized solid-electrolyte interphase layer enables stable lithium metal anodes for long-cycle lithium-sulfur batteries. Nat. Commun. 8, 850 (2017).
Measurement of thin film mechanical properties using nanoindentation. MRS Bull 17, 28–33 (1992).
Viljoen, A. et al. Force spectroscopy of single cells using atomic force microscopy. Nat. Rev. Methods Primers 1 (2021).
Gao, Y. et al. Unraveling the mechanical origin of stable solid electrolyte interphase. Joule 5, 1860–1872 (2021).
Wei, Q. et al. An ultrahigh-power mesocarbon microbeads|Na+ -diglyme|Na3V2(PO4)3 sodium-ion battery. Adv. Mater. 34, e2108304 (2022).
Tai, R.-Z. & Zhao, Z.-T. Overview of SSRF phase-II beamlines. Nucl. Sci. Tech. 35 (2024).
Zhang, L. et al. The 3D nanoimaging beamline at SSRF. Nucl. Sci. Tech. 34 (2023).
Acknowledgements
This work is supported by National Natural Science Foundation of China (Grant Nos. U21A2075, 52561160145), Zhejiang Provincial Natural Science Foundation of China (Grant Nos. LR23B030003, LR24E020001), Jianbing Science and Technology Key Project of Zhejiang Province (2025C01156), International Cooperation and Exchange Program of the National Natural Science Foundation of China (52261135626), Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2023R01007). The authors acknowledge the Chemistry Instrument Center Department of Chemistry Zhejiang University for providing characterization facility, and the Shanghai Synchrotron Radiation Facility of BL18B beamline for the assistance on transmission X-ray microscopy experiments.
Author information
Authors and Affiliations
Contributions
H.P. proposed the research. H.P., Y.L., and L.W. designed the experiments. Y.L. and X.F. performed the material synthesis, investigation, and data analysis, L.W. performed the Synchrotron 3D nanoscale tomography and analysis, X.F., Y.T., Z.C., and S.J. performed electrochemical and AFM measurements, S.W., Y.-S.H., Y.S., and X.L. discussed the mechanisms, H.P., Y.-S.H., Y.L., and X.F. prepared the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kangning Zhao, Likun Zhu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Fan, X., Wang, L. et al. In situ-formed C-N anchors embedded into Sn-based negative electrodes for long-life Na-ion batteries. Nat Commun 17, 2476 (2026). https://doi.org/10.1038/s41467-026-69319-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69319-x
