
Abstract
Pump-probe spectroscopy is a powerful technique for investigating ultrafast exciton dynamics. However, developing a theoretical framework for modeling the transient response in photoexcited materials has remained a challenge. Here, we present a first-principles approach based on a non-equilibrium extension to the Bethe-Salpeter equation to simulate pump-probe spectroscopy and disentangle electronic and thermal contributions to the transient response. Applied to three prototypical semiconductors, i.e., the transition-metal dichalcogenide WSe2, the metal halide perovskite CsPbBr3, and the transition-metal oxide TiO2, the method obtains the transient spectra in excellent agreement with experiment. Our analysis reveals that distinct renormalization mechanisms shape the spectral shifts: Photoinduced Coulomb screening, as the dominant electronic effect, drives excitonic blueshifts, while Pauli blocking plays a minor role. Thermal effects induce redshifts on the picosecond time scale. We further demonstrate how key parameters, such as carrier population distribution, pump wavelength, and pump polarization, impact the transient absorption spectra, which offer direct control over the exciton resonance energy. Our approach establishes a framework for interpreting and tailoring pump-probe spectra, providing guidelines for exciton engineering and thus contributing to the design of energy-selective optoelectronic devices.
Similar content being viewed by others

Introduction
Pump-probe spectroscopy is a state-of-the-art time-resolved technique for investigating light-matter interaction, particularly non-equilibrium processes like exciton dynamics1. In a typical pump-probe absorption experiment, the material is optically excited by a pump pulse, and the resulting transient changes in transmission are measured with a time-delayed probe pulse, uncovering information about exciton dynamics on ultrafast timescales2. Advances in laser technology enable this technique to access a broad range of timescales and energy ranges, from attosecond to picosecond regimes1,3,4, and from the infrared and visible5 to the extreme ultraviolet (XUV)6 and X-ray domains7.
Pump-probe experiments often face limitations in resolving distinct dynamical processes in periodic systems, due to complex many-body interactions. This motivates the development of theoretical methods for simulating transient phenomena. Early theoretical descriptions of transient absorption (TA) spectra were formulated within density-matrix theory8,9,10,11, relying on model Hamiltonians and thus lacking first-principles accuracy. Beyond these approaches, real-time time-dependent density-functional theory (RT-TDDFT) is widely used as a first-principles method to simulate TA spectra12,13,14. While efficient and thus applicable to large systems, RT-TDDFT within a mean-field framework lacks an explicit treatment of many-body interactions. In contrast, many-body wavefunction approaches, such as the time-dependent configuration interaction singles approach15 and coupled cluster theory16, offer a high-accuracy description of electron correlation. However, they are computationally quite involved and thus limited to small atomic or molecular systems. The Bethe-Salpeter equation (BSE) of many-body perturbation theory offers an accurate treatment of electron-hole interaction. It is regarded as the state-of-the-art approach for computing absorption spectra in equilibrium17. However, the effect of photoexcited carriers is not included in standard implementations. Later, this formalism was generalized to the non-equilibrium regime, validated on a simple four-band model system18, and subsequently used to simulate the transient reflectivity spectrum of silicon19. More recently, we took the first step towards treating such pump-probe spectra from first principles, which allowed us to quantify how photoexcited carriers impact the TA spectra in the wide band-gap material ZnO20.
In this work, we present an approach integrating RT-TDDFT, constrained density-functional theory (cDFT), and a non-equilibrium extension to BSE that considers both electronic and thermal contributions fully ab initio. We apply our method to three prototypical materials: tungsten diselenide (WSe2), cesium lead bromide (CsPbBr3), and anatase titanium dioxide (TiO2). These compounds are representative of transition-metal dichalcogenides (TMDCs), metal halide perovskites, and transition-metal oxides, respectively, and are widely used in photodetection21, photocatalysis22,23, and solar energy conversion24,25. We disentangle the spectral features into electronic and thermal contributions, revealing how each modulates the exciton resonance. Our analysis not only enables us to qualitatively interpret and understand available experimental TA spectra, but also predicts how the transient response can be controlled via the carrier population and pump parameters. Our work thus provides a powerful tool for interpreting pump-probe experiments and gaining insight into transient response, offering guidelines for the design of energy-selective optoelectronic devices.
Results and Discussion
First-principles formalism to simulate TA spectra
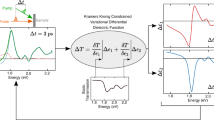
Figure 1 presents the computational workflow used to simulate pump-probe spectra and to disentangle the electronic and thermal contributions. This approach consists of five main steps: computing the ground state, determining photoexcited carrier distributions and expanded lattice structure, solving the equilibrium and non-equilibrium BSE, evaluating the core-level response, and separating the transient response into electronic and thermal contributions. The left panel of Fig. 1 illustrates the optical pump–X-ray probe scheme, while the right panel outlines the computational procedure adopted in this work. Within this framework, the pump-induced non-equilibrium electronic effects are described using either RT-TDDFT or cDFT, depending on the timescale. The pump-induced thermal effect is modeled within density functional theory (DFT) by considering a thermally expanded lattice structure. The probe response is simulated by the BSE formalism. The computational workflow is summarized as follows:

Left: Scheme of the pump-probe process. A pump pulse photoexcites the semiconductor, initiating transitions from valence-band (VB) to conduction-band (CB) states (purple arrow). A time-delayed pulse subsequently probes the transient response of the material by promoting core electrons to CB states (pink arrow). Right: Computational workflow for disentangling electronic and thermal contributions to the TA spectra. fs and ps denote femtosecond and picosecond timescales, respectively. cDFT and NE BSE denote the constrained density functional theory and non-equilibrium Bethe-Salpeter equation methods, respectively. Pauli and Screening represent the photoinduced Pauli blocking and Coulomb screening effects. The blue (red) workflow indicates the procedure to isolate the electronic (thermal) contribution.
• Step 1: DFT ground-state. We first perform a self-consistent DFT calculation to obtain the Kohn-Sham (KS) eigenvalues and orbitals \(\{{\varepsilon }_{n{\bf{k}}}^{0},{\psi }_{n{\bf{k}}}^{0}\}\), which serve as the input for the equilibrium and non-equilibrium BSE calculations.
• Step 2: Photoexcited carrier distribution via two routes.
RT-TDDFT for the femtosecond timescale
The time evolution of photoexcited carrier distributions at femtosecond time delays following optical pumping is simulated using RT-TDDFT. The pump field is modeled through a time-dependent vector potential A(t) (velocity gauge), yielding the time-dependent KS Hamiltonian26:
where VKS(r, t) is the time-dependent KS potential, and c is the speed of light. Starting from the initial condition \({\psi }_{n{\bf{k}}}({\bf{r}},t=0)={\psi }_{n{\bf{k}}}^{0}({\bf{r}})\), the KS orbitals evolve according to the time-dependent KS equations:
To obtain time-dependent photoexcited carrier distributions, the propagated orbitals are projected onto the ground-state KS eigenstates {\({\psi }_{c{\bf{k}}}^{0}\), \({\psi }_{v{\bf{k}}}^{0}\)}. The conduction-band (c) occupations are computed as:
and the valence-band (v) occupations are obtained accordingly,
where \({f}_{n{\bf{k}}}^{0}\) denotes the ground-state KS occupations.
cDFT for the picosecond timescale
At picosecond time delays following the optical pumping, the photoexcited carrier distributions are modeled using cDFT. In the semiconductors investigated in this work (WSe2, CsPbBr3, TiO2), carrier-carrier and carrier-phonon scattering lead to rapid thermalization on sub-picosecond timescales27,28,29. Therefore, on the picosecond timescale, electrons and holes have already relaxed to the vicinity of the conduction-band minimum (CBM) and valence-band maximum (VBM), respectively, such that the photoexcited carrier distribution can be described by Fermi-Dirac distributions characterized by an electronic temperature T:
Here, kB is the Boltzmann constant, μ is the Fermi-level energy in equilibrium, and \({f}_{c{\bf{k}}}^{e}\) (\({f}_{v{\bf{k}}}^{h}\)) denotes the electron (hole) occupation numbers in the conduction (valence) states. The excited electron and hole populations are constrained to satisfy charge neutrality and to match the total number of photoexcited carriers per unit cell, Nexc:
where wk are the weights of the k-points in the Brillouin zone.
• Step 3: Non-equilibrium BSE. The electron-hole wavefunctions are formulated in the basis of ground-state KS orbitals, i.e., the exciton wavefunctions are expanded as
with the expansion coefficients \({A}_{vc{\bf{k}}}^{\lambda }\). The BSE is written as an eigenvalue problem for an effective two-particle Hamiltonian, according to the standard equilibrium formulation30:
with excitation energies Eλ. The BSE Hamiltonian is decomposed as:
where Hdiag describes independent-particle transitions, Hx contains the repulsive exchange interaction mediated by the bare Coulomb potential \(v({\bf{r}},{\bf{r}}{\prime} )\), and Hdir accounts for the attractive electron-hole interaction, which contains the statically screened Coulomb potential \({W}_{{\bf{G}}{{\bf{G}}}^{{\prime} }}({\bf{q}})\). It incorporates the inverse dielectric function, ϵ−1, at ω = 0,
where the microscopic dielectric function, ϵ−1, is calculated within the random-phase approximation (RPA) from the independent-particle polarizability χ0. We evaluate χ0 by replacing the equilibrium carrier distribution with the photoexcited carrier distribution obtained in Step 2:
where η is a positive infinitesimal and \({M}_{vc}^{{\bf{G}}}({\bf{k}},{\bf{q}})=\langle {\psi }_{v{\bf{k}}}^{0}| {e}^{i({\bf{q}}+{\bf{G}})\cdot {\bf{r}}}| {\psi }_{c,{\bf{k+q}}}^{0}\rangle\) are the plane-wave matrix elements.
• Step 4: Core-level response. In the optical limit (q → 0), the macroscopic dielectric function εM(ω) is obtained as31:
where Ω is the unit-cell volume, and tλ(q → 0) is the transition coefficient:
with \(\widehat{p}\) being the momentum operator. From the complex dielectric function, εM(ω), both the absorption α(ω) and the reflectivity coefficients R(ω), can be evaluated32.
• Step 5: Electronic and thermal contribution to the TA spectra.
Electronic contribution
To quantify the electronic contribution to TA spectra, we solve the non-equilibrium BSE using the photoexcited carrier distribution obtained in Step 2, yielding a non-equilibrium absorption spectrum αneq(ω, t). The electronic TA spectrum at a pump-probe delay t is defined as the difference between the non-equilibrium and equilibrium spectra αeq(ω) as
It contains contributions from two many-body effects: (i) Pauli blocking, arising from occupation-induced suppression of optical transitions, and (ii) photoinduced Coulomb screening, i.e., the modification of the electron-hole interaction due to the screening by photoexcited carriers. We disentangle the two contributions by selectively introducing the photoexcited carrier distribution either in the transition coefficients through the factor \(\sqrt{{f}_{v{\bf{k}}}-{f}_{c,{\bf{k+q}}}}\) (Step 4, Eq. (14)) to isolate Pauli blocking, or in the RPA polarizability used to construct the statically screened Coulomb interaction (Step 3, Eq. (11)-(12)) to isolate photoinduced Coulomb screening.
Thermal contribution
At picoseconds time delays after photoexcitation, energy relaxation mediated by exciton-phonon scattering has brought the electronic and lattice subsystems into thermal equilibrium, resulting in an approximately homogeneous lattice temperature. Therefore, the lattice response can be approximated by a spatially uniform thermal expansion33. We model pump-induced heating by constructing a structure with an expanded lattice at an elevated temperature T, and compute the spectra using equilibrium BSE on top of it. Note that exciton-phonon coupling can also modify the spectra, which is not explicitly included here. Accordingly, the thermal contribution to the TA spectrum is approximated as
Finally, we note that the approach presented here is not a formal realization of non-equilibrium BSE derived on the Keldysh contour. A fully non-equilibrium BSE treatment would require explicit two-time propagation34, whose computational cost scales unfavorably with the number of bands, k-points, and time steps, making it impractical for realistic materials35. To enable pump-probe simulations at an affordable cost, we have adopted three approximations. First, the adiabatic approximation assumes that the pump-driven system is frozen during the probe process, and the optical response at each pump-probe delay is evaluated from an instantaneous non-equilibrium carrier distribution18. Second, electronic and thermal effects are separated based on different timescales. Third, thermal effects are approximated by lattice expansion. This approach thus has its limitations: the lack of real-time propagation in the BSE prevents the description of the real-time formation, evolution, and relaxation of excitons. The absence of explicit electron-phonon interaction prevents the description of exciton-phonon scattering. Treating the screening at the static RPA level neglects the dynamical screening, memory effects, and vertex corrections. Despite these limitations, the present approach provides a computationally tractable way to simulate and predict the pump-probe spectra, offering an interpretation of the physical origin of the transient spectral features.
Transient absorption at the Se M edge in WSe2
TMDCs are layered materials whose two-dimensional monolayers are weakly bound by van der Waals forces36,37,38. They exhibit strong photoluminescence, spin-control capabilities, and efficient light absorption. Most of the remarkable properties of TMDCs originate from tightly bound excitons. Therefore, understanding exciton dynamics is essential for advancing TMDC-based optoelectronic and photonic devices36,39,40. Pump-probe TA spectroscopy has revealed that exciton dynamics in TMDCs are highly sensitive to factors such as exciton density and dielectric environment41,42. However, the microscopic mechanisms underlying these observations remain elusive, highlighting the need for first-principles investigations. Here, we focus on WSe2 as a representative TMDC, due to its oxidation stability, high carrier mobility, and ambipolar characteristics43,44, and perform first-principles calculations of TA spectra. To consider the electronic effect, RT-TDDFT and cDFT calculations adopt the pump parameters and excitation densities reported in experiment27 to simulate the photoexcited carrier distribution. To model thermal effects, we choose 700 K as a conservative upper bound that amplifies thermal effects for a clean separation from the electronic counterpart, while remaining below the air-oxidation threshold of 773 K45.
In a first step, we discuss the XUV absorption spectra at the Se M4,5 edge in equilibrium. As observed in Fig. 2a, b, the calculated spectrum (black curve) agrees well with the measured counterpart (shaded gray area), reproducing the two-peak structure and the relative peak positions. The additional measured feature at around 54 eV originates from transitions involving the W O2 edge, which is not included in the calculations. Importantly, the excitonic effects are essential to achieve this level of agreement, since the independent-particle approximation (IPA) cannot even qualitatively describe the peak structure observed in the experiment (Fig. S1). This finding contradicts earlier claims that IPA can yield good qualitative agreement27,46.

a Equilibrium (black curve) and non-equilibrium absorption (yellow/green curve) at femtoseconds, reflecting electronic contributions for different excitation densities ne. b Non-equilibrium absorption on the picosecond scale, showing the electronic effect (blue curve) at an excitation density of 1.0 × 1020 cm−3 and thermal contribution (red curve) at a lattice temperature Tl of 700 K. c, d TA spectra corresponding to a and b, respectively. Experimental spectra (gray area) from ref. 27 are shown for comparison. e, f Decomposition of the electronic TA spectra into photoinduced Coulomb screening (blue area) and Pauli blocking (red area) at femtosecond and picosecond delays. Insets: Zoom into the Pauli blocking effect. g Electron (red circles) and hole (blue circles) distributions in k-space, shown for localized, intermediate, and delocalized carrier populations at a fixed excitation density of 1.0 × 1020 cm−3. h Corresponding TA spectra computed using the carrier distributions shown in g. The spectra are shown in terms of the absorption coefficient, α in a and b, and differential absorption, Δα in c–f and h.
Figure 2a, b show the electronic (yellow, green, and blue curves) and thermal (red curve) contributions to the non-equilibrium absorption spectra on the femtosecond and picosecond timescales. The electronic effect arises from photoexcited carriers at different excitation densities (ne), while the thermal effect reflects the impact of lattice expansion at 700 K. Both contributions renormalize exciton resonance energies: photoexcited carriers induce a blueshift that increases with excitation density, while lattice expansion results in a redshift. The changes relative to the equilibrium spectra are shown in Fig. 2c, d. At a delay of 4 fs, photoexcited carriers dominate the TA spectra. The calculated spectrum at ne = 2.0 × 1019 cm−3 (yellow curve in Fig. 2a) reproduces the experimental features between 55 ~ 56.5 eV (shaded area) very well. In contrast, the peaks observed below 55 eV and above 56.5 eV are likely to be attributed to thermal effects, similarly to what we show for the picosecond timescale (red curve in Fig. 2d). The TA amplitude grows non-linearly with excitation density, since a 15-fold increase in ne does not yield a proportional increase in spectral intensity. At a delay of 5 ps, lattice heating becomes important, as evidenced by the good agreement between the calculated signal near 55 eV and experiment (Fig. 2d).
Furthermore, we disentangle the electronic TA spectra into contributions from Pauli blocking and Coulomb screening by photoexcited carriers, as illustrated in Fig. 2e, f. Pauli blocking (red area) plays a minor role on both the femtosecond and picosecond timescales. Conversely, photoinduced Coulomb screening (blue area) accounts for nearly all features in the TA spectra. By weakening the effective Coulomb interaction between core holes and valence electrons, the exciton binding energies are reduced, giving rise to a spectral blueshift.
Finally, we examine how the TA spectra depend on the carrier distribution in k-space. For a fixed excitation density of 1.0 × 1020 cm−3, Fig. 2g displays the localized, intermediate, and delocalized carrier distribution in k-space. The corresponding TA spectra are depicted in Fig. 2h, indicating that a more delocalized distribution of excited electrons and holes across k-space leads to enhanced screening. This, in turn, amplifies the TA response, with positive and negative features becoming more pronounced. The effect tends to saturate at the intermediate carrier distribution in k-space. This result shows that carrier delocalization in k-space directly determines the strength of screening and the amplitude of the TA signal.
Transient absorption at the Br K-edge in CsPbBr3
Metal halide perovskites have garnered significant interest over the past decade, owing to their unique properties, such as high carrier mobility, tunable band gaps, and high power-conversion efficiencies47,48. These excellent properties extend their broad applications in next-generation optoelectronic devices, including photovoltaics, light-emitting diodes, and photodetectors49. While the performance of such devices has advanced rapidly, it is now approaching theoretical limits47,50. Therefore, understanding the microscopic mechanisms of exciton dynamics remains in great demand for device optimization.
X-ray transient absorption (XTA) with element specificity offers simultaneous sensitivity to both electronic and lattice degrees of freedom, providing valuable insights into excitonic behavior51. Despite its great potential, achieving ultrafast XTA measurement remains technically challenging. This highlights the importance of first-principles calculations, which can interpret and predict XTA spectra, providing practical guidelines for experimental design. Here, we focus on the all-inorganic perovskite CsPbBr3, owing to its high photoluminescence quantum yield and exceptional stability52. We disentangle the electronic and thermal contributions to its XTA spectra using our first-principles approach. Pump parameters for the RT-TDDFT simulations and excitation densities for the cDFT calculations are obtained from experimental works53,54,55. The thermal expansion is evaluated at 350 K, i.e., below the phase transition that occurs around 360 K56.
We first calculate the Br K-edge absorption spectrum in equilibrium, depicted in Fig. 3a, b (black curve), which shows good agreement with experiment (shaded gray area, ref. 53). Upon incorporating the effect of photoexcited carriers, the spectrum experiences a blueshift that increases with excitation density (yellow and green curves). The XTA amplitude shown in Fig. 3c increases nonlinearly with the excitation density. At a delay of 4 fs, it is approximately an order of magnitude larger than that on the picosecond timescale (Fig. 3d), owing to the higher excitation density shortly after the pump pulse. The electronic XTA signal (yellow, green, and blue curves in Fig. 3c, d) reproduces most experimental features, except for the initial positive peak around ~ 13.466 keV, which we assign to thermal effects, as discussed below.

a Equilibrium (black curve) and non-equilibrium absorption (yellow/green curve) at femtoseconds, reflecting electronic contributions for different excitation densities ne. b Non-equilibrium absorption on the picosecond scale, showing the electronic effect (blue curve) at an excitation density of 6.0 × 1018 cm-3 and thermal contribution (red curve) at a lattice temperature Tl of 350 K. c, d TA spectra corresponding to a, b, respectively. Experimental spectra (gray area) from ref. 53 are shown for comparison. e, f Decomposition of the electronic TA spectra into photoinduced Coulomb screening (blue area) and Pauli blocking (red area) at femtosecond and picosecond delays. Insets: Zoom into the Pauli blocking effect. g Electron (red circles) and hole (blue circles) distributions in k-space, shown for localized, intermediate, and delocalized carrier populations at a fixed excitation density of 6.0 × 1018 cm-3. h Corresponding TA spectra computed using the carrier distributions shown in (g).
Thermal expansion at 350 K induces a renormalization of the exciton resonance at a time delay of 1 ps, manifested as a redshift in the absorption spectra (red curve in Fig. 3b). This shift originates from the reduced energy separation between the Br 1s core level and the conduction bands. To date, femtosecond XTA experiments on CsPbBr3 have not been reported. Our first-principles investigation can predict the XTA response on a short timescale for guiding future femtosecond XTA experiments.
Figure 3e, f show different effects contributing to the electronic components of the XTA spectra, for both the femtosecond and picosecond timescales. The dominant effect is Coulomb screening by excited carriers (blue area). The impact of Pauli blocking (red area) plays a minor role; it nevertheless leads to a positive signal in the pre-edge region around ~ 13.468 keV, corresponding to core transitions to depopulated orbitals, as well as a negative peak around ~ 13.473 keV, resulting from blocked transitions to occupied states. Photoinduced Coulomb screening reduces electron-hole Coulomb interactions, decreases exciton binding energies, and is responsible for the blueshifted non-equilibrium absorption spectra in Fig. 3a, b. These results demonstrate how photoexcited carriers manipulate Coulomb screening. We conclude that precise control over excitonic resonance energies can be achieved by adjusting the excitation density.
We further investigate the effect of photoexcited carrier distributions on the Coulomb screening by employing Fermi-Dirac statistics at different electronic temperatures in Fig. 3g, while keeping the excitation density fixed at 6.0 × 1018 cm−3. The corresponding XTA spectra in Fig. 3h reveal that a more delocalized distribution in k-space lead to a larger XTA amplitude, indicating enhanced screening. This effect saturates at the intermediate carrier distribution in k-space, consistent with our observations in WSe2. These results demonstrate that in this material, not only excitation density but also carrier distribution can modulate photoinduced Coulomb screening and exciton resonance energies.
Transient absorption at the Ti K-edge in anatase TiO2
Anatase TiO2 is one of the most promising metal-oxide semiconductors for photocatalysis57. Upon photoexcitation, electron-hole pairs are created: electrons at Ti sites drive reduction reactions, while holes at O sites mediate oxidation. XTA spectroscopy can track changes in the oxidation state and the local atomic structure, which can be used to identify the active sites and guide the design of effective catalysts58. However, only a few experiments have targeted XTA spectra at the Ti K-edge, due to the considerable technical demands of high-energy X-ray measurements58,59. Also here, we can bridge the gap between experiment and theory, providing predictive insight into the XTA spectra at the Ti K-edge of anatase TiO2. Pump parameters for RT-TDDFT and excitation densities for cDFT calculations are taken from experimental works59,60. Thermal effects are evaluated at 800 K, well below the phase transition of TiO2 occurring around 870 K61.
The equilibrium absorption spectrum at the Ti K-edge is depicted in Fig. 4a, b, showing good agreement with the experimental counterpart (shaded gray area, ref. 58). We first examine the electronic contributions to the absorption spectra (yellow, green, and blue curves). The non-equilibrium spectra at both femtosecond and picosecond time delays exhibit a blueshift, which is more pronounced in the femtosecond case, due to the higher excitation density. This carrier-induced blueshift produces a negative signal near 4.984 keV in the XTA spectra (Fig. 4c), consistent with experiment58. We next analyze the thermal contributions arising from lattice expansion at 800 K and a time delay of 1 ps (red curve in Fig. 4b). In this case, a redshift of the exciton resonance occurs, originating from the reduced energy separation between the Ti 1s core level and the conduction band. This thermal effect produces a positive peak around 4.982 keV in the XTA spectrum (Fig. 4d), a feature also observed in experiment, but not reproduced when considering the electronic effect alone. This thermal contribution is scaled by a factor of 0.2 for comparison with the electronic TA spectra.

a Equilibrium (black curve) and non-equilibrium absorption (yellow/green curve) at femtoseconds, reflecting electronic contributions for different excitation densities ne. b Non-equilibrium absorption on the picosecond scale, showing the electronic effect (blue curve) at an excitation density of 1.8 × 1019 cm-3 and thermal contribution (red curve) at a lattice temperature Tl of 800 K. c, d TA spectra corresponding to a, b, respectively. Experimental spectra (gray area) from ref. 58 are shown for comparison. e, f Decomposition of the electronic TA spectra into photoinduced Coulomb screening (blue area) and Pauli blocking (red area) at femtosecond and picosecond delays. Insets: Zoom into the Pauli blocking effect. g Electron (red circles) and hole (blue circles) distributions in k-space, shown for localized, intermediate, and delocalized carrier populations at a fixed excitation density of 1.8 × 1019 cm-3. h Corresponding TA spectra computed using the carrier distributions shown in (g).
Like the other materials, we disentangle the effects that shape the transient response in Fig. 4e, f. Photoinduced Coulomb screening (blue area) dominates the XTA spectra across both timescales. In contrast, Pauli blocking (red area) plays a comparatively minor role. Its influence is more noticeable at early femtosecond delays, consistent with our findings at the Br K-edge in CsPbBr3. Likewise, with increasing excitation density, the enhanced photoinduced Coulomb screening further decreases the exciton binding energy (Fig. S2), leading to a blueshift of the exciton resonance in the absorption spectra (Fig. 4a, b). These results highlight Coulomb screening as the key mechanism modulating exciton resonance in TiO2 and demonstrate its universality across different material classes.
We further investigate the role of carrier distribution using Fermi-Dirac statistics at different electronic temperatures for a fixed excitation density of 1.8 × 1019 cm−3 (Fig. 4g). The corresponding XTA spectra are shown in Fig. 4h. The delocalization of carrier distribution in k-space enhances dielectric screening, thereby weakening the Coulomb interaction between electrons and holes. A progressively more delocalized carrier distribution gives rise to an increasing XTA amplitude. This increase saturates at the intermediate carrier distribution in k-space. The overall behavior is consistent with our observations in WSe2 and CsPbBr3.
Beyond TA spectra, we further investigate the electronic contribution to the transient reflectivity (TR) spectra on both femtosecond and picosecond timescales. As shown in Fig. 5a, b, electronic excitations induce a blue shift in the spectrum, which becomes increasingly prominent at higher excitation densities, all concomitant to the behavior of the TA spectra. Obviously, this shift arises primarily from photoinduced Coulomb screening (Fig. 5e, f). In contrast, the thermal contribution to the spectra (red curve in Fig. 5b) leads to a red shift relative to the equilibrium response. The presence of delocalized carrier distributions further amplifies the response in the TR spectra (Fig. 5g, h), as observed also for the TA spectra. The TR spectra for WSe2 (Se M4,5-edge) and CsPbBr3 (Br K-edge) are provided in Fig. S3-S4 in the Supplementary Note 4.1.

a Equilibrium (black curve) and non-equilibrium reflectivity (yellow/green curve) at femtoseconds, reflecting electronic contributions for different excitation densities ne. b Non-equilibrium reflectivity on the picosecond scale, showing the electronic effect (blue curve) at an excitation density of 1.8 × 1019 cm-3 and thermal contribution (red curve) at a lattice temperature Tl of 800 K. c, d TR spectra corresponding to a, b, respectively. e, f Decomposition of the electronic TR spectra into photoinduced Coulomb screening (blue area) and Pauli blocking (red area) at femtosecond and picosecond delays. Insets: Zoom into the Pauli blocking effect. g Electron (red circles) and hole (blue circles) distributions in k-space, shown for localized, intermediate, and delocalized carrier populations at a fixed excitation density of 1.8 × 1019 cm-3. h Corresponding TR spectra computed using the carrier distributions shown in g. The spectra are shown in terms of the reflectivity coefficient, R in (a, b), and differential reflectivity, ΔR in (c-f) and (h).
Control of photoinduced Coulomb screening and exciton resonance
For the three materials studied so far, we have observed that higher excitation densities and more delocalized carrier distributions in k-space enhance photoinduced Coulomb screening. Beyond these two parameters, we consider now the polarization direction of the pump beam. We take anatase TiO2 as an example, as it exhibits a well-known anisotropic dielectric response, with stronger absorption when the electric field aligns with the z-direction62. At a delay of 20 fs, our simulations show that a pump polarization oriented along the x-direction yields a smaller excitation density compared to the polarization along z (Fig. S5a). In both cases, the degree of delocalization in k-space is similar (Fig. 6a). The higher excitation density induced by a z-polarized pump gives rise to a stronger XTA signal (Fig. 6b), indicating stronger Coulomb screening. This finding opens the possibility for tuning exciton resonances through the polarization of the pump beam.

Time evolution of photoexcited electron (red circles) and hole (blue circles) distributions following femtosecond optical excitation with varying a pump polarizations (x and z), and c pump wavelengths (355 nm and 266 nm). b, d Corresponding transient absorption (TA) spectra.
The pump wavelength is another degree of freedom to explore, since it may also affect exciton resonances. Exciting anatase TiO2 with different wavelengths at the same pump fluence results in nearly the same excitation densities (Fig. S5b), yet with a more pronounced XTA spectrum for the shorter wavelength (Fig. 6d). The excitation with shorter wavelength populates the electronic states over a broader energy range in k-space, also further away from the band edges (Fig. 6c), which increases their polarizability and consequently enhances the dielectric screening. These findings identify the pump wavelength as an additional control parameter for tuning exciton resonance energies.
Controlling exciton screening in view of modulating exciton resonances offers broad opportunities for optoelectronic applications. Such control enables the tuning of the detection windows of X-ray detectors without altering the device structure. It also allows control over the energy range contributing to second-harmonic generation (SHG), thereby enabling the modulation of the generated second-harmonic wavelength. Overall, controlling exciton screening enables tunable light-matter interaction across a broad spectral range and is promising for the design of energy-selective photonic devices, such as photodetectors, nonlinear optical devices, and optical filters.
In summary, we have developed an approach that combines RT-TDDFT and cDFT with the non-equilibrium extension to the BSE to simulate pump-probe spectra and disentangle electronic and thermal contributions to the TA spectra. The present implementation relies on several controlled approximations, including an adiabatic treatment of photoexcited states, a timescale-based separation of electronic and thermal effects, and an approximate treatment of thermal effects by lattice expansion. These approximations enable the simulation of the pump-probe spectra in realistic materials at an affordable computational cost. We have applied this method to three prototypical materials, i.e., WSe2, CsPbBr3, and anatase TiO2, achieving excellent agreement with experiments.
Our results reveal that thermal effects caused by lattice heating induce redshifts of exciton resonances. Photoinduced Coulomb screening dominates the electronic effects that reduce exciton binding energies, thus resulting in blueshifts of the spectra. Pauli blocking is a minor electronic effect. Moreover, we have demonstrated that exciton resonance energies can be tuned through excitation density, carrier distribution, pump polarization, and wavelength. These insights provide practical guidelines for tailoring optical parameters to design energy-selective optoelectronic devices.
The ab initio approach presented here becomes a reference scheme for simulating pump-probe spectra across a wide range of materials. It can capture the dominant mechanisms governing transient response across timescales, ranging from attoseconds to picoseconds and energy ranges, spanning the visible and near-infrared to extreme ultraviolet and X-ray regimes. This capability is crucial for identifying future targets for attosecond core-level transient spectroscopy, which remains challenging to achieve experimentally. This approach is also well-suited for modeling transient reflectivity spectra. Looking ahead, it can be extended to simulate other time-resolved probes, such as time-resolved terahertz spectroscopy and time-resolved photoluminescence.
Methods
Ground-state calculations
All calculations were performed using the all-electron, full-potential package exciting. Brillouin zone integrations were performed using a Monkhorst-Pack k-point grid of 18 × 18 × 5 for WSe2, 6 × 6 × 4 for CsPbBr3, and 8 × 8 × 6 for TiO2. The Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was employed to describe exchange-correlation effects. Scalar relativistic effects, including the mass-velocity and Darwin terms, were treated using the atomic ZORA (zero-order regular approximation) scheme, while spin-orbit coupling was neglected.
Excited-state calculations
Table 1 shows the computational parameters employed in the cDFT, RT-TDDFT, and BSE calculations for WSe2, CsPbBr3, and TiO2. The k-point meshes define the Brillouin-zone sampling, whereas \({R}_{MT}| {\bf{G}}+{\bf{k}}{| }_{\max }\) determines the LAPW basis-set cutoff. In RT-TDDFT, the pump photon energy (Ehv), polarization, and pulse duration specify the frequency, orientation, and temporal profile of the external field. In the BSE calculations, \({| {\bf{G}}+{\bf{q}}| }_{\max }\) defines the cutoff for the KS response function, dielectric screening, and the Coulomb-potential expansion. The broadening defines the Lorentzian linewidth applied to the spectra. Nempty determines the unoccupied orbitals included in the calculation of screening and electron-hole interaction.
All spectra were computed within the BSE formalism using Kohn-Sham eigenvalues and eigenvectors from ground-state DFT calculations. Since the G0W0 approach is insufficient to yield reliable quasiparticle corrections for core states63, we adopted a scissor correction by aligning the calculated equilibrium spectra with the experimental counterparts. The scissor correction has been widely employed in previous studies of core excitations, and is generally sufficient to describe the spectral lineshape and relative peak positions64,65. Given that the main objective of our work is to resolve pump-induced transient spectra, i.e., the difference between equilibrium and non-equilibrium spectra, this approach is justified. The screened Coulomb interaction W entering the BSE kernel was evaluated without scissor correction, in line with standard BSE practice.
Data Availability
All input and output files are available in the NOMAD data infrastructure with the following link: https://doi.org/10.17172/NOMAD/2025.09.01-1. exciting code for all simulations is available at https://exciting-code.org/.
References
Ashoka, A. et al. Extracting quantitative dielectric properties from pump-probe spectroscopy. Nat. Commun. 13, 1437 (2022).
Beeby, A.An Introduction to Laser Spectroscopy, chap. Pump-probe laser spectroscopy, 105–137 2 edn. (Springer, Boston, MA., 2002).
Lucchini, M. et al. Unravelling the intertwined atomic and bulk nature of localised excitons by attosecond spectroscopy. Nat. Commun. 12, 1021 (2021).
Barends, T. R. et al. Influence of pump laser fluence on ultrafast myoglobin structural dynamics. Nature 626, 905–911 (2024).
Montanaro, A. et al. Visible pump–mid infrared pump–broadband probe: Development and characterization of a three-pulse setup for single-shot ultrafast spectroscopy at 50 khz. Rev. Sci. Instrum. 91 (2020).
Rebholz, M. et al. All-xuv pump-probe transient absorption spectroscopy of the structural molecular dynamics of di-iodomethane. Phys. Rev. X 11, 031001 (2021).
Geneaux, R., Marroux, H. J., Guggenmos, A., Neumark, D. M. & Leone, S. R. Transient absorption spectroscopy using high harmonic generation: a review of ultrafast x-ray dynamics in molecules and solids. Philos. Trans. R. Soc., A 377, 20170463 (2019).
Lindberg, M. & Koch, S. Theory of coherent transients in semiconductor pump–probe spectroscopy. J. Opt. Soc. Am. B 5, 139–146 (1988).
Pollard, W. T., Lee, S.-Y. & Mathies, R. A. Wave packet theory of dynamic absorption spectra in femtosecond pump–probe experiments. J. Chem. Phys. 92, 4012–4029 (1990).
Yan, Y. J., Zhang, W. & Che, J. Time–frequency theory of pump-probe absorption spectroscopy. J. Chem. Phys. 106, 2212–2224 (1997).
Wolfseder, B., Seidner, L., Stock, G. & Domcke, W. Femtosecond pump-probe spectroscopy of electron-transfer systems: a nonperturbative approach. Chem. Phys. 217, 275–287 (1997).
De Giovannini, U., Brunetto, G., Castro, A., Walkenhorst, J. & Rubio, A. Simulating pump–probe photoelectron and absorption spectroscopy on the attosecond timescale with time-dependent density functional theory. ChemPhysChem 14, 1363–1376 (2013).
Pemmaraju, C. Simulation of attosecond transient soft x-ray absorption in solids using generalized kohn–sham real-time time-dependent density functional theory. New J. Phys. 22, 083063 (2020).
Moitra, T., Konecny, L., Kadek, M., Rubio, A. & Repisky, M. Accurate relativistic real-time time-dependent density functional theory for valence and core attosecond transient absorption spectroscopy. J. Phys. Chem. lett. 14, 1714–1724 (2023).
Pabst, S. et al. Theory of attosecond transient-absorption spectroscopy of krypton for overlapping pump and probe pulses. Phys. Rev. A: At. Mol. Opt. Phys. 86, 063411 (2012).
Skeidsvoll, A. S., Balbi, A. & Koch, H. Time-dependent coupled-cluster theory for ultrafast transient-absorption spectroscopy. Phys. Rev. A 102, 023115 (2020).
Onida, G., Reining, L. & Rubio, A. Electronic excitations: density-functional versus many-body green’s-function approaches. Rev. Mod. Phys. 74, 601 (2002).
Perfetto, E., Sangalli, D., Marini, A. & Stefanucci, G. Nonequilibrium bethe-salpeter equation for transient photoabsorption spectroscopy. Phys. Rev. B 92, 205304 (2015).
Sangalli, D., Dal Conte, S., Manzoni, C., Cerullo, G. & Marini, A. Nonequilibrium optical properties in semiconductors from first principles: A combined theoretical and experimental study of bulk silicon. Phys. Rev. B 93, 195205 (2016).
Rossi, T. C. et al. Dynamic control of x-ray core-exciton resonances by coulomb screening in photoexcited semiconductors. Commun. Mater. 6, 191 (2025).
Peng, J. et al. Crystallization of CsPbBr3 single crystals in water for X-ray detection. Nat. Commun. 12, 1531 (2021).
Zheng, Z. et al. Flexible, transparent and ultra-broadband photodetector based on large-area WSe2 film for wearable devices. Nanotechnology 27, 225501 (2016).
Yu, B. et al. Enhanced photocatalytic properties of graphene modified few-layered WSe2 nanosheets. Appl. Surf. Sci. 400, 420–425 (2017).
Lewerenz, H., Heller, A. & DiSalvo, F. Relationship between surface morphology and solar conversion efficiency of tungsten diselenide photoanodes. J. Am. Chem. Soc. 102, 1877–1880 (1980).
Ullah, S. et al. All-inorganic CsPbBr3 perovskite: a promising choice for photovoltaics. Mater. Adv. 2, 646–683 (2021).
Yabana, K., Sugiyama, T., Shinohara, Y., Otobe, T. & Bertsch, G. F. Time-dependent density functional theory for strong electromagnetic fields in crystalline solids. Phys. Rev. B 85, 045134 (2012).
Oh, J. et al. Carrier and phonon dynamics in multilayer WSe2 captured by extreme ultraviolet transient absorption spectroscopy. J. Phys. Chem. C 127, 5004–5012 (2023).
Chen, J., Messing, M. E., Zheng, K. & Pullerits, T. Cation-dependent hot carrier cooling in halide perovskite nanocrystals. J. Am. Chem. Soc. 141, 3532–3540 (2019).
Park, S. H. et al. Direct and real-time observation of hole transport dynamics in anatase TiO2 using X-ray free-electron laser. Nat. Commun. 13, 2531 (2022).
Rohlfing, M. & Louie, S. G. Electron-hole excitations and optical spectra from first principles. Phys. Rev. B 62, 4927–4944 (2000).
Albrecht, S., Reining, L., Del Sole, R. & Onida, G. Ab initio calculation of excitonic effects in the optical spectra of semiconductors. Phys. Rev. Lett. 80, 4510–4513 (1998).
Ambrosch-Draxl, C. & Sofo, J. O. Linear optical properties of solids within the full-potential linearized augmented planewave method. Comput. Phys. Commun. 175, 1–14 (2006).
Bauerhenne, B. & Garcia, M. E. Unified description of thermal and nonthermal laser-induced ultrafast structural changes in materials. Sci. Rep. 14, 32168 (2024).
Keldysh, L. V. Diagram technique for nonequilibrium processes. Soviet Physics—JETP 20, 1018–1026 (1965).
Tuovinen, R., Covito, F. & Sentef, M. A. Efficient computation of the second-born self-energy using tensor-contraction operations. J. Chem. Phys. 151 (2019).
Ji, H. et al. Thickness-dependent carrier mobility of ambipolar MoTe2: Interplay between interface trap and Coulomb scattering. App. Phys. Lett. 110 (2017).
Niu, Y. et al. Thickness-dependent differential reflectance spectra of monolayer and few-layer MoS2, MoSe2, WS2 and WSe2. Nanomaterials 8, 725 (2018).
Kolobov, A. V. & Tominaga, J.Bulk TMDCs: Review of Structure and Properties, 29–77 (Springer International Publishing, Cham, 2016).
Britz, A. et al. Carrier-specific dynamics in 2H-MoTe2 observed by femtosecond soft x-ray absorption spectroscopy using an x-ray free-electron laser. Struct. Dyn. 8, 014501 (2021).
Duong, N. T. et al. Modulating the functions of MoS2/MoTe2 van der Waals heterostructure via thickness variation. ACS nano 13, 4478–4485 (2019).
Pöllmann, C. et al. Resonant internal quantum transitions and femtosecond radiative decay of excitons in monolayer WSe2. Nat. Mater. 14, 889–893 (2015).
Raja, A. et al. Coulomb engineering of the bandgap and excitons in two-dimensional materials. Nat. Commun. 8, 15251 (2017).
Liu, W. et al. Role of metal contacts in designing high-performance monolayer n-type WSe2 field effect transistors. Nano Lett. 13, 1983–1990 (2013).
Wang, Z. et al. The ambipolar transport behavior of WSe2 transistors and its analogue circuits. NPG Asia Mater. 10, 703–712 (2018).
Zou, Y. et al. High-temperature flexible WSe2 photodetectors with ultrahigh photoresponsivity. Nat. Commun. 13, 4372 (2022).
Schumacher, Z. et al. Ultrafast electron localization and screening in a transition metal dichalcogenide. Proc. Natl. Acad. Sci. 120, e2221725120 (2023).
National Renewable Energy Laboratory. Best research-cell efficiency chart Accessed: 2025-07-13 (2024).
Thakur, D. & Chang, S. H. Material properties and optoelectronic applications of lead halide perovskite thin films. Synth. Met. 301, 117535 (2024).
Wu, S., Chen, Z., Yip, H.-L. & Jen, A. K.-Y. The evolution and future of metal halide perovskite-based optoelectronic devices. Matter 4, 3814–3834 (2021).
Shockley, W. & Queisser, H. J. Detailed balance limit of efficiency of p–n junction solar cells. J. Appl. Phys. 32, 510–519 (1961).
Chen, L. X., Zhang, X. & Shelby, M. L. Recent advances on ultrafast x-ray spectroscopy in the chemical sciences. Chem. Sci. 5, 4136–4152 (2014).
Zhang, M. et al. Stable CsPbBr3 perovskite quantum dots with high fluorescence quantum yields. New J. Chem. 42, 9496–9500 (2018).
Cannelli, O. et al. Quantifying photoinduced polaronic distortions in inorganic lead halide perovskite nanocrystals. J. Am. Chem. Soc. 143, 9048–9059 (2021).
Lv, J. et al. Hot carrier trapping and it’s influence to the carrier diffusion in CsPbBr3 perovskite film revealed by transient absorption microscopy. Adv. Sci. 11, 2403507 (2024).
Polishchuk, S. et al. Nanoscale-resolved surface-to-bulk electron transport in CsPbBr3 perovskite. Nano Lett. 22, 1067–1074 (2022).
Mannino, G. et al. Temperature-dependent optical band gap in CsPbBr3, MAPbBr3, and FAPbBr3 single crystals. J. Phys. Chem. Lett. 11, 2490–2496 (2020).
Regan, O. & Gratzel, M. Light induced charge separation in nanocrystalline films. Nature 353, 737–740 (1991).
Santomauro, F. G. et al. Femtosecond X-ray absorption study of electron localization in photoexcited anatase TiO2. Sci. Rep. 5, 14834 (2015).
Rittmann-Frank, M. H. et al. Mapping of the photoinduced electron traps in TiO2 by picosecond x-ray absorption spectroscopy. Angew. Chem., Int. Ed. 53, 5858–5862 (2014).
Baldini, E., Palmieri, T., Pomarico, E., Aubock, G. & Chergui, M. Clocking the ultrafast electron cooling in anatase titanium dioxide nanoparticles. ACS Photonics 5, 1241–1249 (2018).
Hanaor, D. A. & Sorrell, C. C. Review of the anatase to rutile phase transformation. J. Mater. Sci. 46, 855–874 (2011).
Kang, W. & Hybertsen, M. S. Quasiparticle and optical properties of rutile and anatase TiO2. Phys. Rev. B:Condens. Matter Mater. Phys. 82, 085203 (2010).
Li, J., Jin, Y., Rinke, P., Yang, W. & Golze, D. Benchmark of GW methods for core-level binding energies. J. Chem. Theory Comput. 18, 7570–7585 (2022).
Cocchi, C. & Draxl, C. Bound excitons and many-body effects in x-ray absorption spectra of azobenzene-functionalized self-assembled monolayers. Phys. Rev. B 92, 205105 (2015).
Vorwerk, C., Cocchi, C. & Draxl, C. Addressing electron-hole correlation in core excitations of solids: An all-electron many-body approach from first principles. Phys. Rev. B 95, 155121 (2017).
Acknowledgements
Financial support from the Alexander von Humboldt Foundation and the German Research Foundation, projects 424709454 and 182087777 are acknowledged. This work was performed using HPC resources from “Lise’’ at the NHR center NHR@ZIB. The authors acknowledge support from the Open Access Publication Fund of Humboldt-Universität zu Berlin. L.Q. acknowledges M. Benedikt for valuable discussions. The authors acknowledge support by the Open Access Publication Fund of Humboldt-Universität zu Berlin.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.Q. conceptualized the work and proposed the theoretical approach, performed the calculations for CsPbBr$_3$ and TiO$_2$, and wrote the manuscript. R.R.P. implemented the RT-TDDFT and cDFT methods, carried out the calculations for WSe$_2$, and wrote the corresponding discussion. R.R.P. and C.D. reviewed and revised the manuscript. All authors contributed to the discussion of the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qiao, L., Pela, R.R. & Draxl, C. First-principles Approach to Ultrafast Pump-probe Spectroscopy in Semiconductors. npj Comput Mater 12, 179 (2026). https://doi.org/10.1038/s41524-026-02128-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-026-02128-4
