
Abstract
Ectonucleotidases, including NTPDases and ecto-5′-nucleotidase (e-5′NT/CD73), regulate extracellular purinergic signaling by converting ATP to adenosine, a pathway critically involved in immune response, inflammation, and cancer progression. In this study, a novel library of 22 N-propylsulfonyl-substituted indole-based hydrazinecarbothioamides (5a–5v) was synthesized and structurally characterized. Biological evaluation against human e-5′NT and NTPDase1, -2, -3, and − 8 revealed that several compounds exhibited low micromolar inhibitory activity, with 5n (IC50 = 1.7 µM), 5o (IC50 = 1.7 µM), 5f (IC50 = 1.0 µM), and 5i (IC50 = 1.6 µM) emerging as the most promising derivatives, showing strong potency and isoform selectivity. Structure–activity relationship analysis indicated that both electronic and steric features of substituents significantly influence activity and enzyme preference. Molecular docking studies performed on e-5′NT demonstrated that active compounds adopt consistent binding modes within the catalytic pocket, stabilized by key residues such as Asp-506, Phe-500, Phe-417 and Arg-395. Binding free energy calculations (MM-GBSA) supported strong ligand–protein interactions ( ~ − 70 kcal/mol). The docking protocol was validated by redocking, yielding an RMSD value well below the accepted threshold. Molecular dynamics simulations (500 ns) confirmed stable complex formation, with low RMSD values (~ 1–3 Å), limited residue fluctuations, and persistent interactions with catalytic residues. Surface and compactness parameters (rGyr, SASA) remained stable, indicating consistent ligand accommodation. In silico ADME analysis suggested favorable drug-like properties for most compounds, particularly for the lead candidates. Overall, these findings identify 5n and 5o as the most promising lead compounds, supported by both experimental and computational results, and highlight this scaffold as a valuable platform for the development of selective ectonucleotidase inhibitors.
Similar content being viewed by others

Introduction
Ectonucleotidases are a family of metal-dependent enzymes that regulate extracellular nucleotide levels by catalyzing the sequential hydrolysis of nucleoside tri-, di-, and monophosphates. In this pathway, ecto-5′-nucleotidase (e-5′NT) converts AMP to adenosine, while upstream enzymes such as NTPDases hydrolyze ATP and ADP to AMP. Based on their catalytic functions, ectonucleotidases are classified into four main groups: ecto-nucleoside triphosphate diphosphohydrolases (NTPDases), ecto-nucleotide pyrophosphatases/phosphodiesterases (NPPs), alkaline phosphatases (APs), and ecto-5′-nucleotidase (e-5′NT)1,2,3,4,5,6.
The ecto-NTPDase family comprises eight isoforms (NTPDase1–8), among which NTPDase1, −2, −3, and − 8 are membrane-bound and directly involved in extracellular nucleotide hydrolysis, exhibiting distinct substrate preferences (e.g., NTPDase1 hydrolyzes both ATP and ADP, whereas NTPDase2 preferentially hydrolyzes ATP). Together with ecto-5′-nucleotidase (e-5′NT), a zinc-dependent enzyme that converts AMP to adenosine, these enzymes form a coordinated cascade regulating ATP-to-adenosine conversion at the cell surface7,8,9,10,11. Increased expression of CD39 (NTPDase1) and CD73 (e-5′NT) in hypoxic tumor environments leads to elevated adenosine production, which is associated with tumor progression and immune evasion12,13,14,15.
In the tumor microenvironment, elevated extracellular ATP is rapidly converted into adenosine through the coordinated action of CD39 (NTPDase1) and CD73 (e-5′NT). The resulting accumulation of adenosine plays a key role in suppressing antitumor immune responses by inhibiting T-cell activation and promoting immune evasion. This pathway has therefore emerged as a critical immune checkpoint mechanism in cancer progression. Consequently, inhibition of ectonucleotidases, particularly CD73 and NTPDases, represents a promising strategy to restore immune function and enhance anticancer therapy16,17,18.
Ectonucleotidases, particularly CD39 (NTPDase1) and CD73 (e-5′NT), have emerged as important therapeutic targets due to their central role in regulating extracellular adenosine levels16,19,20,21. Overactivation of this pathway leads to excessive adenosine accumulation, which suppresses immune responses and promotes tumor growth, angiogenesis, and metastasis, making these enzymes key targets in cancer immunotherapy. Beyond oncology, dysregulated purinergic signaling has also been implicated in inflammatory diseases, autoimmune disorders, and cardiovascular conditions, where abnormal nucleotide/adenosine balance contributes to disease progression. Therefore, inhibition of ectonucleotidases represents a promising strategy to restore extracellular nucleotide homeostasis and modulate disease-associated signaling pathways as illustrated in Fig. 1.

Ectonucleotidase (CD39/CD73) pathway and its inhibition: regulation of ATP-adenosine balance and its importance in immunosuppression and disease progression.
The indole moiety is a great scaffold for developing drug -like moleules with anticancer potential. The indole ring is a well-established biologically active scaffold capable of interacting with a wide range of drug targets22,23,24,25. Indole-based compounds occupy a prominent position in anticancer drug discovery26,27,28. Notably, clinically approved indole-containing chemotherapeutics such as vincristine and vinblastine exert their antitumor activity by disrupting microtubule dynamics, leading to mitotic arrest and apoptosis in rapidly dividing cancer cells. The proven clinical efficacy of these agents has motivated continued efforts to design and synthesize novel indole derivatives with enhanced potency, selectivity, and improved pharmacokinetic and safety profiles, thereby addressing limitations such as toxicity, resistance, and suboptimal bioavailability associated with existing therapies29,30,31.
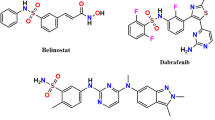
Thiosemicarbazones (TSCs) are a class of Schiff bases recognized for their diverse pharmacological activities and strong metal-chelating properties31. Numerous TSC derivatives have demonstrated promising antitumor activity32,33. in in-vitro assays against a variety of cancers, including leukemia and solid tumors such as pancreatic, prostate, breast, cervical, and bladder cancers34,35,36,37,38,39,40,41,42,43,44. Figure 2 illustrates selected inhibitors of ectonucleotidases that have been reported in the literature17,18,45,46,47,48.

Reported structures of NTPDase and e-5′NT inhibitors along with current work.
Drug design
From a synthetic perspective, tethering two or more biologically active moieties is a popular trend and an effective therapeutic strategy in medicinal chemistry49. This pharmacophore hybridization process offers several advantages, such as reduced drug resistance and improved efficacy compared to the parent moieties. This concept is not only limited to developing multi-target agents, but rather focuses on combining complementary structural motifs to enhance potency, selectivity, physicochemical behavior, or potential to overcome drug resistance.
In the current work, the two well-established pharmacophore moieties, the indole core and the thiosemicarbazone (TSC) moiety, are integrated into a single molecular framework. This hybridization approach was designed to combine the proven utility of the indole scaffold in anticancer drug discovery with the metal-chelating and biologically active properties of TSCs, aiming to generate molecules with enhanced inhibitory activity against ectonucleotidases. The present work represents a structure-activity relationship (SAR) extension of previously reported N-hexylsulfonyl indole-based thiosemicarbazones bearing the TSC moiety at the C-3 position17. We sought to investigate the effect of relocating the thiosemicarbazone functionality to the C-5 position. Accordingly, a new series of N-propylsulfonyl-substituted indole-based hydrazinecarbothioamides was synthesized and evaluated for inhibitory activity against NTPDase1, NTPDase2, NTPDase3, NTPDase8, and ecto-5′-nucleotidase (e-5′NT).
Results and Discussion
Chemistry
Given the numerous biological and pharmacological activities associated with indole and thiosemicarbazone skeletons, a combination of these scaffolds into a single molecular framework to synthesize more powerful compounds, such as 5(a–v), with better biological properties is logical. The target molecules, i.e., N-substituted indole-based thiosemicarbazones, have been obtained in two steps. In the first step, Indole-5-carbaldehyde (1) is substituted at the N position by the reaction of indole-5-carbaldehyde with propyl sulfonyl chloride in the presence of DMAP, and triethylamine using DCM as a solvent, which leads to the replacement of the H of indole-5-carbaldehyde at the NH position with the propyl group. After extraction using a sodium bicarbonate solution, the desired 1-(propylsulfonyl)indole-5-carbaldehyde (3) was produced as a white precipitate with a remarkable yield of 85–95%.
In the second step, the obtained compound (3) is reacted with various thiosemicarbazides 4(a-v) by refluxing an equimolar mixture of both in ethanol, using a catalytic amount of acetic acid, to yield a library of N-substituted indole-based TSCs 5(a–v) at the C-5 position. The synthetic route for the TSCs 5(a–v) is shown in Fig. 3.

Schematic representation of the synthesis of thiosemicarbazones 5(a-v).
FT-IR, 1H & 13C NMR, HPLC, and ESI-MS results verified the synthesis of N-substituted indole intermediate (3) and TSCs 5(a–v). The appearance of two singlets for NH protons at 12.19–11.52 ppm and 10.50–9.07 ppm indicates that thiosemicarbazide was successfully condensed at the indole’s aldehydic site, as well as a singlet for the azomethine proton at 8.57–8.17 ppm in the 1H NMR spectra of representative TSCs 5(a–v). The propyl group that attaches to the indole ring at N position is responsible for the signals in the aliphatic region between 0.84 and 3.77 ppm. C = S and C = N are liable for the peaks in the 13C NMR at 178.13–175.76 and 162.62-142.59.62.59 ppm, respectively. The N-H band in FT-IR for thiosemicarbazones 5(a-v) appears between 3117 and 3369 cm− 1. Within the range of 2933 to 29,779 cm− 1, aromatic C-H emerged. The development of a novel azomethine C = N bond in TSCs was confirmed concurrently by the appearance of CN extending from 1502 to 1573 cm− 1. The observed range of the thiourea moiety’s C = S band was 1121–1217 cm− 1. In ESI spectra, the molecular ion peak matched the targeted substances’ molecular mass exactly. The aforementioned peaks from many methods verified that the desired thiosemicarbazones were formed.
For the fluorinated derivative 5o, the C–F coupling analysis was performed to confirm the exact position of the fluorine atom on the aromatic ring using characteristic C–F coupling patterns. The results are consistent with literature data50,51,52,53. The C–F coupling analysis of compound 5o (see Supporting Figure) confirms the fluorine substitution pattern. The ipso carbon (C21, δ = 161.99 ppm) shows a strong 1J_C–F coupling (241.7 Hz), while the ortho carbons (C22 and C20) exhibit typical 2J_C–F values (24.5 and 21.0 Hz). The meta carbons (C17 and C19) display 3J_C–F couplings (10.9 and 9.4 Hz), and the para carbon (C18) shows a weak long-range 4J_C–F coupling (2.8 Hz). This pattern is consistent with a fluorinated phenyl ring and confirms the correct positioning of the fluorine substituent. Similar coupling behavior was observed for the other fluorinated derivatives.
The stereochemistry of the thiosemicarbazone C = N bond is proposed to be predominantly E based on spectroscopic and computational evidence. The 1H NMR spectra display a single set of signals, particularly a characteristic azomethine proton singlet, indicating the presence of one major isomer in solution and excluding a significant E/Z mixture at the NMR timescale. This observation is consistent with literature reports, where thiosemicarbazones generally favor the E configuration due to reduced steric hindrance. In addition, MM2 (ChemDraw 3D) calculations indicate that the E isomer is energetically more stable than the Z isomer (ΔE ≈ 1.44 kcal/mol). However, in the absence of definitive experimental techniques such as X-ray crystallography or NOESY analysis, this assignment should be considered tentative.
In vitro enzyme inhibition studies
The target compounds were evaluated against e-5′NT and NTPDase1, −2, −3, and − 8, revealing several highly potent inhibitors with low micromolar IC50 values, surpassing standard inhibitors, and the results are given in Table 1. Especially target compounds include 5n (1.7 µM, e-5′NT), 5i (1.6 µM, NTPDase1), 5f (1.1 µM, NTPDase2; 1.0 µM, NTPDase8), and 5o (1.7 µM, NTPDase3; 1.1 µM, NTPDase8). Several compounds also showed marked isoform selectivity, underscoring the scaffold’s potential for developing targeted enzyme inhibitors.
Structure activity relationship (SAR)
The structure–activity relationship (SAR) analysis of the synthesized thiosemicarbazones revealed clear and isoform-dependent trends, highlighting the critical role of substituent type, position, and steric compatibility in determining both potency and selectivity. Across all isoforms, substitution at the phenyl ring was essential for activity, as the unsubstituted analogue 5a was inactive or less active in most cases. However, the nature and position of substituents governed distinct and sometimes opposing preferences depending on the enzyme isoform.
For h-e5′NT and NTPDase3, electron-withdrawing groups (EWGs), particularly halogens, significantly enhanced activity, especially when positioned at the meta position. Representative examples include 5n (o-CF3) and 5o (m-F) for e5′NT, and 5o (m-F), 5 L (3,5-bis-CF3), and 5s (2,6-diCl) for NTPDase3. These results suggest that these enzymes favor substituents that provide both electronic withdrawal and optimal spatial orientation, enabling better accommodation within relatively constrained binding pockets. Importantly, not all EWGs improved activity, indicating that steric fit and substituent orientation are more decisive than electronic effects alone.
In contrast, NTPDase2 and NTPDase8 displayed a distinct preference for electron-donating groups (EDGs), particularly at the para position. Compound 5f (p-methyl) emerged as the most potent inhibitor for both isoforms (IC50 = 1.1 µM for NTPDase2 and 1.0 µM for NTPDase8), indicating that hydrophobic and electron-donating substituents enhance binding affinity in these enzymes. This suggests that these active sites are more hydrophobic and sterically permissive, allowing favorable interactions with para-oriented substituents.
NTPDase1 exhibited a unique SAR profile, where disubstitution patterns were more influential than simple electronic effects. The most potent compound, 5i (2,4-dimethyl), along with 5t (2,6-difluoro), demonstrated that specific substitution patterns can optimize both steric complementarity and interaction geometry. In contrast, increasing steric bulk beyond an optimal level (e.g., benzyl in 5d or trityl in 5v) led to reduced activity, confirming that steric hindrance limits access to the active site.
Selectivity trends further support these observations. For example, compound 5f showed strong selectivity toward NTPDase2 and NTPDase8, while 5o displayed multi-target activity with high potency toward NTPDase3 and NTPDase8. Similarly, compounds such as 5s and 5p exhibited selective inhibition toward NTPDase3, indicating that substituent positioning (meta vs. ortho vs. para) is a key determinant of isoform selectivity.
Overall, the SAR analysis demonstrates that enzyme selectivity is governed by a balance of electronic effects, steric compatibility, and substituent orientation. While EWGs favor h-e5′NT and NTPDase3, EDGs, particularly at the para position, enhance inhibition of NTPDase2 and NTPDase8. NTPDase1, on the other hand, requires specific disubstitution patterns for optimal activity. These findings provide a coherent mechanistic framework for the rational design of selective ectonucleotidase inhibitors. The observed structure–activity relationship trends are summarized in Fig. 4 and are further supported by docking analysis.

Illustration of structure structure-activity relationship of the synthesized compounds with h-e-5’NT and NTPDase isoforms.
Molecular docking studies and MM-GBSA analysis
To investigate the molecular basis of the observed inhibitory activity, molecular docking studies were focused on ecto-5′-nucleotidase (e-5′NT, CD73). This target was selected based on the availability of a human X-ray crystal structure in the Protein Data Bank (PDB), enabling reliable structure-based analysis. In contrast, other ectonucleotidase isoforms lack experimentally determined crystal structures and would require homology modeling. Therefore, docking studies were performed only on e-5′NT using six compounds that exhibited biological activity.
Accordingly, molecular docking studies were performed on all six compounds that exhibited biological activity, namely 5n (2-trifluoromethylphenyl), 5o (3-fluorophenyl), 5 g (cyclohexyl), 5k (3-methoxyphenyl), 5j (4-bromo-2-fluorophenyl), and 5q (4-chlorophenyl). These compounds were selected based on their experimentally observed activity, while the remaining compounds were inactive and therefore not included in the docking analysis. Docking was performed using the Induced Fit Docking (IFD) protocol (accounting for receptor flexibility and ligand-induced conformational changes), and binding affinities were further evaluated by MM-GBSA (Molecular Mechanics/Generalized Born Surface Area, ΔG_bind) calculations (post-docking binding free energy estimation combining molecular mechanics energies with implicit solvation and surface area terms). The obtained results are presented in Table 2.
Molecular docking results for the six active compounds against ecto-5′-nucleotidase (e-5′NT) showed a narrow range of IFD docking scores (− 7.200 to − 7.839 kcal/mol), indicating similar binding orientations within the active site. Among them, compound 5n exhibited the most favorable docking score (− 7.839 kcal/mol), while 5q showed the least favorable value.
In contrast, MM-GBSA binding free energy calculations provided clearer discrimination between the compounds. Compounds 5n (− 71.94 kcal/mol), 5 g (− 71.21 kcal/mol), and 5o (− 71.05 kcal/mol) demonstrated the most favorable binding energies, suggesting more stable protein–ligand complexes. Compound 5k showed moderate affinity (− 61.70 kcal/mol), whereas 5q was weaker (− 56.08 kcal/mol). Notably, compound 5j exhibited a significantly less favorable MM-GBSA value (− 32.14 kcal/mol) despite a comparable docking score, indicating reduced stability after solvation and energy refinement.
MM-GBSA energy decomposition analysis
To gain deeper insight into the energetic contributions governing ligand binding, MM-GBSA energy decomposition analysis was performed. This approach enables the dissection of the total binding free energy (ΔG_bind) into individual components, including Coulombic (electrostatic), covalent, hydrogen bonding, lipophilic, solvation (GB), and van der Waals (vdW) interactions. Such decomposition provides a more detailed understanding of the dominant forces stabilizing the ligand–protein complexes and helps to identify the key interaction types contributing to binding affinity and selectivity54,55. The results are presented in Table 3.
According to the Table 3, van der Waals (vdW) and lipophilic interactions were the main favorable contributors to binding, indicating that hydrophobic interactions play a dominant role in ligand stabilization within the active site. In contrast, the polar solvation term (Solv GB) generally contributed unfavorably, partially offsetting the binding energy. Among the compounds, 5n and 5o exhibited strong vdW contributions (− 56.58 and − 56.41 kcal/mol, respectively), supporting their stable binding profiles observed in docking and MD analyses. Compound 5j showed a more favorable electrostatic contribution (Coulomb, − 6.17 kcal/mol), suggesting a relatively stronger role of polar interactions. Overall, the balance between hydrophobic (vdW/lipophilic) and polar (Coulomb/solvation) terms governs the binding affinity of the compounds.
Based on these results, compounds 5n and 5o were selected for further in-depth analysis. Their ligand–protein interactions were evaluated in detail. The ligand–protein interactions (LPI) of compounds 5o and 5n are presented as 2D and 3D representations in Fig. 4.
The 2D interaction diagram of 5n (Fig. 4a) reveals a rich interaction network within the active site. The thiosemicarbazone nitrogen forms a hydrogen bond with Asp-506, while the imine nitrogen establishes an additional hydrogen bond with Asn-390. The indole ring participates in three π–π stacking interactions with Phe-500 and Phe-417 (two interactions), indicating strong aromatic stabilization. In addition, the trifluoromethyl-substituted phenyl ring engages in a π–cation interaction with Arg-395. Similarly, the 2D LPI profile of 5o (Fig. 4c) shows a comparable binding pattern. The indole ring forms three π–π stacking interactions with Phe-417 and Phe-500, consistent with 5n. The imine nitrogen interacts with Asn-390 via hydrogen bonding, while the thiosemicarbazide nitrogen forms another hydrogen bond with Asp-506. These results highlight the importance of key residues such as Asp-506, Asn-390, Phe-417, Phe-500, and Arg-395 in ligand stabilization.

Molecular docking LPI analysis of 5n and 5o against e5’-NT. (a) 2D LPI of 5n (b) 3D LPI of 5n, (c) 2D LPI of 5o (b) 3D LPI of 5o.
The 3D interaction views (Fig. 4b and d) further support these findings. In these representations, yellow dashed lines indicate hydrogen bonds, cyan dashed lines represent π–π stacking interactions, and red dashed lines denote π–cation interactions. The bluish surface corresponds to the ligand binding surface, whereas the grey surface represents the protein binding pocket. A high degree of surface overlap between these regions indicates an optimal fit within the active site. Both 5n and 5o exhibit excellent surface complementarity with minimal solvent exposure of the phenyl moieties. Quantitatively, the hydrogen bond distances are highly similar for both compounds (5n: 1.87 and 2.24 Å; 5o: 1.86 and 2.26 Å), indicating comparable interaction strength. The π–π stacking distances range from 3.79 to 4.31 Å for 5n and 3.81–4.27 Å for 5o, further confirming similar aromatic interactions. Additionally, the π–cation interaction observed for 5n occurs at a distance of 6.30 Å.
As a result, both compounds exhibit highly similar binding modes and interaction patterns within the active site. However, compound 5n demonstrates slightly enhanced binding interactions, particularly due to the additional π–cation interaction with Arg-395, which may contribute to its higher biological activity compared to 5o. Nevertheless, 5o also shows a well-preserved interaction network and strong binding stability, indicating that both compounds are effective binders of e-5′-NT.
Molecular dynamics (MD) simulation studies
To further validate the stability and dynamic behavior of the ligand–protein complexes, MD simulations were performed for the 5n-e-5’NT and 5o-e-5’NT complexes over a 500 ns timescale. The MD trajectories were analyzed using key parameters such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (Rg) to evaluate the structural stability, flexibility, and compactness of the complexes. These parameters are essential to determine whether the system reaches equilibrium and maintains a stable conformation over time. In addition, solvent-accessible surface area (SASA) and hydrogen bond analyses were considered to monitor ligand exposure to the solvent and the persistence of intermolecular interactions, which are critical for stable binding56. The MD simulation analyses of the 5n–e-5′NT and 5o–e-5′NT complexes are presented in Figs. 5, 6 and 7.
Figure 5 presents the 500 ns MD simulation results of the 5n–e-5’NT complex. In Fig. 5a, key ligand-protein interactions observed throughout the simulation are illustrated. The two nitrogen atoms of the thiourea moiety within the thiosemicarbazide group formed persistent hydrogen bond interactions with Asp-506 for 95% of the simulation time. The imine nitrogen of the TSC group established water-bridged hydrogen bonds with Arg-395 (32% of the sim.) and Gly-392 (30% of the sim.). Phe-500 contributed to binding via dual interactions, forming hydrogen bonds with the sulfonyl oxygen (69% of the sim.) and π–π stacking interactions with the indole ring (30% of the sim.). Additionally, the indole ring engaged in π–π stacking with Phe-417 at a higher frequency (61% of the sim.). The trifluorophenyl ring exhibited a strong π–cation interaction with Arg-441 (71% of the sim.).

500 ns MD simulation of the 5n–e-5’NT complex: (a) key 2D ligand–protein interactions; (b) RMSD profiles of protein Cα atoms (pale blue) and ligand (red), including ligand positional deviation (pink); (c) RMSF of protein residues with ligand-contact regions highlighted; (d) RMSF of ligand atoms; (e) interaction fraction histogram showing hydrogen bond and hydrophobic contact frequencies over the trajectory.
Figure 5b shows the RMSD profiles. The protein Cα atoms remained stable with an average RMSD of 1.4 Å (light blue), while the ligand RMSD relative to the protein (ligand fit on protein) averaged 3.0 Å (red). In contrast, the ligand internal RMSD (ligand fit on ligand) was lower, with an average of 1.2 Å (pink), indicating limited conformational changes. Figure 5c and d present RMSF profiles of protein Cα atoms and ligand atoms, respectively. In Fig. 5c, green vertical lines indicate residues maintaining contacts with the ligand during the simulation. The average RMSF values were 0.9 Å for protein residues and 1.25 Å for ligand atoms, suggesting only minor fluctuations within the expected dynamic range.
Finally, Fig. 5e displays the interaction fraction histogram. In MD simulations, a residue may simultaneously interact with multiple functional groups of ligand, and conversely, a single functional group may interact with multiple residues. Therefore, fractional interaction values are cumulative and may exceed 1.0. The y-axis represents the total interaction fraction over the trajectory, where green indicates hydrogen bonds, blue represents water-bridged hydrogen bonds, and purple corresponds to hydrophobic interactions. According to the histogram, the most persistent interacting residues are Asp-506, Phe-500, and Phe-417.
Figure 6 presents the 500 ns MD simulation results of the 5o–e-5’NT complex. In Fig. 6a, a binding pattern similar to that of 5n is observed. The TSC nitrogen atoms formed highly persistent hydrogen bond interactions with Asp-506 (97–98%). Additionally, the TSC sulfur atom and imine nitrogen established hydrogen bonds with Arg-395 at frequencies of 44% and 58%, respectively. Phe-500 played a dual role, forming hydrogen bonds with the sulfonyl oxygen (92%) and π–π stacking interactions with the indole ring (67%). The indole ring also interacted with Phe-416 via π–π stacking (63%). Furthermore, the fluorophenyl ring exhibited a π–cation interaction with Arg-441 (32%).
Figure 6b shows the RMSD profiles, where the protein Cα atoms remained stable with an average RMSD of 1.2 Å. The ligand RMSD (ligand fit on protein) averaged 2.4 Å, while the ligand internal RMSD (ligand fit on ligand) was 1.6 Å, indicating moderate but stable conformational adaptation. Figure 6c and d present RMSF profiles of protein and ligand atoms, respectively. The average RMSF values were 0.9 Å for protein residues and 1.25 Å for the ligand, suggesting limited fluctuations within the expected dynamic range. Finally, Fig. 6e shows the interaction fraction histogram. The most prominent and persistent interacting residues were Asp-506, Phe-500, Phe-417, and Arg-395, consistent with the key interactions observed throughout the simulation.

500 ns MD simulation of the 5o–e-5’NT complex: (a) key 2D ligand–protein interactions; (b) RMSD profiles of protein Cα atoms (pale blue) and ligand (red), including ligand positional deviation (pink); (c) RMSF of protein residues with ligand-contact regions highlighted; (d) RMSF of ligand atoms; (e) interaction fraction histogram showing hydrogen bond and hydrophobic contact frequencies over the trajectory.
To provide a more comprehensive view of the dynamic behavior and interaction persistence throughout the simulation, timeline analysis and additional structural parameters are presented in Figs. 7 and 8.

MD simulation timeline analysis and additional structural parameters of 5n– and 5o–e-5’NT complexes. (a–b) Protein–ligand contact timelines for 5n and 5o complexes (500 ns). (c–d) Structural parameters (RMSD, rGyr, MolSA, SASA, PSA) indicating stable binding; no intramolecular H-bonds observed.
The contact timeline analysis reveals that the total number of protein–ligand contacts stabilizes after ~ 100–150 ns for both systems. Residue-wise maps clearly show continuous high-frequency interactions, particularly for Asp-506 and Phe-500, which maintain near-continuous contact throughout the 500 ns trajectory (interaction occupancy 80–100%). Additional residues such as Phe-417 and Arg-395 exhibit intermittent but still significant interaction patterns (40–70% occupancy), supporting their role in stabilizing the ligand within the binding pocket.
The RMSD profiles (d–e, top panels) confirm structural stability, with average values of approximately 1.0–1.3 Å for 5n and 1.2–1.6 Å for 5o, remaining within a narrow fluctuation range (ΔRMSD < 0.5 Å after equilibration). The radius of gyration (rGyr) values are confined to ~ 5.8–6.4 Å, indicating preservation of ligand compactness. Surface-related descriptors further support stable binding behavior. The molecular surface area (MolSA) remains within 395–405 Å2 (5n) and 372–385 Å2 (5o), while SASA values fluctuate within 150–210 Å2, suggesting no major exposure changes. PSA values are also stable, centered around 95–110 Å2, indicating consistent polarity during the simulation. Notably, no intramolecular hydrogen bonds were detected for either ligand, implying that ligand stability is predominantly governed by intermolecular interactions with the protein rather than internal conformational locking. The low variance (σ) in RMSD, rGyr, and surface parameters, together with sustained high contact occupancy, quantitatively confirms that both 5n and 5o form stable and persistent complexes with e-5’NT over the 500 ns simulation.
As a result of the MD simulations, both 5n and 5o maintained stable binding throughout the 500 ns trajectory, supported by low RMSD values (1.2–1.4 Å for protein; 2.4–3.0 Å for ligand) and minimal RMSF fluctuations (0.9–1.25 Å). Key amino acids such as Asp-506, Phe-500, and Phe-417 exhibited high interaction occupancies (up to 95–100%), indicating persistent binding. The rGyr (5.8–6.4 Å), MolSA, SASA, and PSA profiles remained within narrow ranges, confirming compact ligand conformations and consistent surface exposure. These results demonstrate that both compounds form stable and well-maintained complexes with e-5’NT.
In Silico ADME evaluation
The pharmacokinetic properties of the synthesized compounds (5a-v) were evaluated using in silico ADME prediction tools to assess their drug-likeness and oral bioavailability potential. Compounds 5 h and 5u were excluded from the ADME analysis due to their lack of biological activity against all tested enzymes. Key descriptors, including molecular weight (MW), hydrogen bond acceptors (aHB), hydrogen bond donors (dHB), lipophilicity (QPlogPo/w), brain–blood partition coefficient (QPlogBB), aqueous solubility (QPlogS), Caco-2 and MDCK cell permeability (QPPCaco and QPPMDCK), and predicted human oral absorption (%HOA), were calculated. In addition, compliance with Lipinski’s Rule of Five (Ro5) and Jorgensen’s Rule of Three (Ro3) was analyzed to evaluate the suitability of the compounds as potential drug candidates. The ADME prediction results are presented in Table 4.
As indicated in Table 4, the compound series shows a generally consistent and favorable ADME profile. All compounds possess acceptable hydrogen bonding capacities (aHB = 9–10, dHB = 2) and most molecular weights fall within the recommended range, with only 5 L and 5v slightly exceeding typical limits. Lipophilicity values are largely within acceptable boundaries, although higher values (e.g., 5v) may negatively affect solubility.
Permeability results indicate that most compounds exhibit high Caco-2 and MDCK values (> 500), suggesting good intestinal absorption and membrane permeability. An exception is 5b, which shows relatively low permeability and slightly reduced oral absorption. In contrast, the majority of compounds display excellent predicted oral absorption (100%). While some compounds (e.g., 5 L and 5v) show lower solubility, this is consistent with their higher lipophilicity. All compounds comply with Jorgensen’s Rule of Three, with only minor Lipinski deviations observed for 5 L and 5v. Notably, compounds 5n and 5o exhibit well-balanced ADME properties, with suitable molecular weight, lipophilicity, high permeability (QPPCaco and QPPMDCK > 800), and excellent oral absorption (~ 100%). These features, together with their strong binding stability observed in MD simulations, highlight their potential as promising candidates.
Docking validation
Docking validation was performed by redocking the co-crystallized ligand (4-nitrocatechol) into the active site of ecto-5′-nucleotidase (e-5′NT, CD73) (PDB ID: 7PBY; resolution: 1.13 Å). The crystal structure corresponds to the open conformation of the enzyme. The superposition of the co-crystallized ligand (green) and the redocked pose (pink) showed excellent agreement, with an RMSD value of 0.0749 Å (Fig. 9).

Docking validation: superposition of co-crystallized (green) and redocked (pink) ligands.
RMSD values below 2.0 Å are generally considered indicative of a reliable docking protocol, while values between 2.0 and 3.0 Å are still acceptable. The extremely low RMSD value obtained in this study indicates a near-perfect reproduction of the experimental binding mode, confirming the high accuracy and robustness of the docking protocol57,58. Furthermore, key active site residues involved in the binding of the co-crystallized ligand, including Arg-354, Arg-395, Asn-390, Asp-506, and Phe-500, were also consistently observed to interact with the studied compounds.
Materials and methods
General
The chemicals and solvents were acquired from Merck and used as such: DCM, ethanol, methanol, petroleum ether, glacial acetic acid, DMAP, triethylamine, ethyl acetate, and substituted thiosemicarbazides. Utilising silica gel plates, the reaction’s progress and completion were monitored. To obtain 1H and 13C NMR spectra at 25 °C in DMSO-d6, a 500 MHz Bruker Ascend NMR spectrometer was used (1H = 500 MHz and 13C = 125 MHz). To demonstrate signal multiplicity, NMR spectra were presented as chemical shifts (ppm) and coupling constants (J) in Hertz (Hz). The Waters preparative HPLC and PDA detector was used to record HPLC chromatograms. HPLC purity analyses were carried out using a Waters preparative HPLC system equipped with a PDA detector. A C18 reversed-phase column was used for all measurements and maintained at a constant temperature of 25 °C. The chromatographic method was performed with a flow rate of 5 mL/min, an injection volume of 100 µL, and an operating pressure of approximately 150 bar. All purity percentages and retention times (Rt) reported in the study were obtained under these standardized conditions. The electrospray ionisation (ESI) method was used to acquire HRMS spectra using a Thermo Fisher Scientific Q ExactiveTM Hybrid Quadrupole-OrbitrapTM device.
General Method for the Synthesis of 1-(propylsulfonyl)−1H-indole-5-carbaldehyde (3)
To a solution of Indole 5-carbaldehyde (1) (2 mmol, 0.29 g) in DCM (15 mL), triethylamine solution (2.6 mmol, 363 µL), DMAP (2 mmol, 0.245 g), and 2 mmol (0.37 g) of 1-propane sulfonyl chloride (2) were added under an argon atmosphere. The reaction mixture was stirred at room temperature for 12 h. A saturated solution of 10% NaHCO3 (10 mL) was mixed in the reaction mixture. In the next step, the extraction of the reaction mixture was carried out three times with 20 mL of DCM. The collected organic layers were dried under a vacuum using a rotary evaporator. The white residue of the compound (3) was purified by flash chromatography utilizing eluent (1:4 ethyl acetate/hexane)17.
1H-NMR (500 MHz, DMSO-d6) δ: 10.07 (1 H, s), 8.29 (1 H, d, J = 1.6 Hz), 8.02 (1 H, d, J = 8.6 Hz), 7.89 (1 H, dd, J = 8.6, 1.7 Hz), 7.74 (1 H, d, J = 3.7 Hz), 7.02 (1 H, d, J = 3.7 Hz), 3.75–3.59 (2 H, m), 1.52 (2 H, p, J = 7.5 Hz), 0.85 (3 H, t, J = 7.4 Hz); 13C-NMR (125 MHz, DMSO-d6) δ: 192.70, 137.87, 132.03, 130.32, 128.99, 125.28, 124.62, 113.69, 108.62, 55.31, 16.80, 12.15.
The general method for the TSCs 5(a-v) Synthesis
To a 10 mL ethanol solution, equimolar quantities (1 mmol) of N-propyl sulfonyl chloride (0.14 g) (3) and commercially available substituted thiosemicarbazides 4(a-v) were mixed. To catalyze the reaction mixture, 1 mL of CH3COOH was added. The reaction mixture was refluxed for 4–6 h at 70 °C. Using the eluent (1:5 petroleum ether/ethyl acetate), the progress of the reaction was checked by thin-layer chromatography. After completion of the reaction, the reaction mixture was cooled to room temperature and the resulting solid residue was collected by filtration. The crude solid was washed thoroughly with ethanol to remove unreacted starting materials and other soluble impurities. The product was dried under reduced pressure to obtain compounds 5(a–v) in moderate to good yields.
N -phenyl-2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5a
Color: white; M.P.: 199 °C; Yield: 95%; FT-IR (cm− 1): 3315 (N-H), 2976 (Ar. C-H), 1534 (C = N), 1163 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.79 (1 H, s, NH-N = C), 10.09 (1 H, s, S = C-NH-R), 8.27 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.9 Hz, Ar.), 7.97 (1 H, dd, J = 8.8, 1.7 Hz, Ar.), 7.86 (1 H, d, J = 8.7 Hz, Ar.), 7.71–7.54 (3 H, m, Ar.), 7.47–7.34 (2 H, m, Ar.), 7.21 (1 H, dt, J = 8.9, 4.6 Hz, Ar.), 6.86 (1 H, d, J = 3.7 Hz, Ar.), 3.60 (2 H, dd, J = 8.5, 6.5 Hz, SO2-CH2), 1.62–1.46 (2 H, mCH2 propyl), 0.85 (3 H, t, J = 7.3 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 176.03(C═S), 143.35 (C═N), 139.25 (Ar), 135.57 (Ar), 130.43 (Ar), 129.45 (Ar), 128.21 (Ar), 128.19 (Ar), 125.96 (Ar), 125.44 (Ar), 123.93 (Ar), 121.59 (Ar), 113.22 (Ar), 108.17 (Ar), 55.19 (SO2-CH2), 16.82 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.22 min, 99.97%; ESI-HRMS: [M + H]+: Cal. 401.11059; Found: 401.10943.
N -(3-nitrophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-y]methylene)hydrazinecarbothioamide 5b
Color: yellow; M.P.: 155 °C; Yield: 94%; FT-IR (cm− 1): 3314 (N-H), 2979 (Ar. C-H), 1537 (C = N), 1197 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 12.07 (1 H, s, NH-N = C), 10.40 (1 H, s S = C-NH-R), 8.66 (1 H, t, J = 2.2 Hz, Ar.), 8.31 (1 H, s, HC═N), 8.19 (1 H, d, J = 1.6 Hz, Ar.), 8.14 (1 H, ddd, J = 8.1, 2.2, 1.0 Hz, Ar.), 8.05 (1 H, ddd, J = 8.3, 2.3, 1.0 Hz, Ar.), 7.99 (1 H, dd, J = 8.7, 1.6 Hz, Ar.), 7.88 (1 H, d, J = 8.7 Hz, Ar.), 7.69–7.62 (2 H, m, Ar.), 6.88 (1 H, dd, J = 3.7, 0.8 Hz, Ar.), 3.70–3.51 (2 H, m, SO2-CH2), 1.58–1.46 (2 H, m, CH2 propyl), 0.85 (3 H, t, J = 7.4 Hz, CH3 propyl). 13C-NMR (125 MHz, DMSO-d6) δ: 175.89 (C═S), 147.45 (C═N), 144.37 (Ar), 140.50 (Ar), 135.70 (Ar), 131.91 (Ar), 130.43 (Ar), 129.34 (Ar), 129.21 (Ar), 128.29 (Ar), 127.35 (Ar), 124.00 (Ar), 121.79 (Ar), 119.79 (Ar), 119.75 (Ar), 113.26 (Ar), 108.17 (Ar), 55.22 (SO2-CH2), 16.82 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.05 min, 99.98%; ESI-HRMS: [M-H]+ Cal.: 444.08002; Found: 444.08126.
N -(4-bromophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5c
Color: off-white; M.P.: 113 °C; Yield: 92%; FT-IR (cm− 1): 3300 (N-H), 2971 (Ar. C-H), 1532 (C = N), 1194 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.88 (1 H, s, NH-N = C), 10.13 (1 H, s, S = C-NH-R), 8.27 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.6 Hz, Ar.), 7.97 (1 H, dd, J = 8.8, 1.7 Hz, Ar.), 7.86 (1 H, d, J = 8.7 Hz, Ar.), 7.64 (1 H, d, J = 3.6 Hz, Ar.), 7.61–7.58 (2 H, m, Ar.), 7.57–7.53 (2 H, m, Ar.), 6.86 (1 H, dd, J = 3.7, 0.8 Hz, Ar.), 3.67–3.55 (2 H, m, SO2-CH2), 1.60–1.40 (2 H, m, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 175.91(C═S), 143.72 (C═N), 138.69 (Ar), 135.60 (Ar), 131.00 (Ar), 130.41 (Ar), 129.34 (Ar), 128.23 (Ar), 127.87 (Ar), 127.74 (Ar), 123.95 (Ar), 121.65 (Ar), 117.62 (Ar), 113.22 (Ar), 108.15 (Ar), 55.19 (SO2-CH2), 16.81 (CH2 propyl), 12.19 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.18 min, 97.01%; ESI-HRMS: [M-H]+ Cal.: 479.00546; Found: 479.00459.
N-benzyl-2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5d
Color: white; M.P.: 133–135 °C; Yield: 90%; FT-IR (cm− 1): 3144 (N-H), 2970 (Ar. C-H), 1535 (C = N), 1136 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.56 (1 H, s, NH-N = C), 9.07 (1 H, t, J = 6.3 Hz, S = C-NH-R), 8.20 (1 H, s, HC═N), 8.07 (1 H, d, J = 1.6 Hz, Ar.), 7.90–7.81 (2 H, m, Ar.), 7.62 (1 H, d, J = 3.7 Hz, Ar.), 7.39–7.28 (4 H, m, Ar.), 7.26–7.20 (1 H, m, Ar.), 6.84 (1 H, d, J = 3.6 Hz, Ar.), 4.86 (2 H, d, J = 6.2 Hz, CH2 Benzyl), 3.65–3.57 (2 H, m, SO2-CH2), 1.54–1.46 (2 H, m, CH2 propyl), 0.83 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 177.69 (C═S), 142.72 (C═N), 139.64 (Ar), 135.43 (Ar), 130.41 (Ar), 129.62 (Ar), 128.29 (Ar), 128.18 (Ar), 127.35 (Ar), 126.83 (Ar), 123.57 (Ar), 121.22 (Ar), 113.21 (Ar), 108.12 (Ar), 55.16 (SO2-CH2), 46.72 (CH2 benzyl), 16.80 (CH2 propyl), 12.18 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.96 min, 96.06%; ESI-HRMS: [M + H]+ Cal.: 415.12624; Found: 415.12480.
N -(naphthalen-1-yl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5e
Color: yellow, M.P.: 190–192 °C, Yield: 97%; FT-IR (cm− 1): 3315 (N-H), 2966 (Ar. C-H), 1529 (C = N), 1142 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.91 (1 H, s, NH-N = C), 10.41 (1 H, s, S = C-NH-R), 8.32 (1 H, s, HC═N), 8.21 (1 H, d, J = 1.6 Hz, Ar.), 8.02 (1 H, dd, J = 8.8, 1.6 Hz, Ar.), 7.98 (1 H, dd, J = 6.3, 3.3 Hz, Ar.), 7.92–7.86 (2 H, m, Ar.), 7.84 (1 H, d, J = 8.7 Hz, Ar.), 7.63 (1 H, d, J = 3.7 Hz, Ar.), 7.58–7.51 (4 H, m, Ar.), 6.83 (1 H, d, J = 3.7 Hz, Ar.), 3.63–3.58 (2 H, m, SO2-CH2), 1.51 (2 H, p, J = 7.5 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 178.00 (C═S), 143.16 (C═N), 135.93 (Ar), 135.54 (Ar), 133.89 (Ar), 130.82 (Ar), 130.45 (Ar), 129.67 (Ar), 128.22 (Ar), 128.16 (Ar), 127.09 (Ar), 126.67 (Ar), 126.24 (Ar), 126.19 (Ar), 125.60 (Ar), 123.97 (Ar), 123.59 (Ar), 121.58 (Ar), 113.32 (Ar), 113.21 (Ar), 108.17 (Ar), 55.17 (SO2-CH2), 16.83 (CH2 propyl), 12.21 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.92 min, 99.77%; ESI-HRMS: [M-H]+ Cal.: 449.11059; Found: 449.11195.
2-([1-(propylsulfonyl)−1H-indol-5-yl]methylene)-N-(p-tolyl)hydrazinecarbothioamide 5f
Color: yellow; M.P.: 156 °C; Yield: 93%; FT-IR (cm− 1): 3305 (N-H), 2969 (Ar. C-H), 1546 (C = N), 1141 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.75 (1 H, s, NH-N = C), 10.02 (1 H, s, S = C-NH-R), 8.25 (1 H, s, HC═N), 8.18–8.15 (1 H, m, Ar.), 7.97 (1 H, dd, J = 8.8, 1.6 Hz, Ar.), 7.85 (1 H, d, J = 8.7 Hz, Ar.), 7.63 (1 H, d, J = 3.7 Hz, Ar.), 7.44 (2 H, d, J = 8.1 Hz, Ar.), 7.17 (2 H, d, J = 8.0 Hz, Ar.), 6.85 (1 H, d, J = 3.7 Hz, Ar.), 3.64–3.57 (2 H, m, SO2-CH2), 2.31 (3 H, s, CH3), 1.51 (2 H, h, J = 7.5 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 176.12 (C═S), 143.20 (C═N), 136.71 (Ar), 135.56 (Ar), 134.63 (Ar), 130.50 (Ar), 130.44 (Ar), 129.50 (Ar), 128.69 (Ar), 128.22 (Ar), 125.97 (Ar), 123.93 (Ar), 121.56 (Ar), 113.23 (Ar), 108.18 (Ar), 55.19 (SO2-CH2), 20.75 (aliph.), 16.83 (CH2 propyl), 12.21 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.96 min, 99.98%; ESI-HRMS: [M + H]+ Cal.: 415.12624; Found: 415.12473.
N -cyclohexyl-2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5 g
Color: white; M.P.: 152 °C; Yield: 94%; FT-IR (cm− 1): 3154 (N-H), 2933 (Ar. C-H), 1534 (C = N), 1121 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.38 (1 H, s, NH-N = C), 8.17 (1 H, s, HC═N), 8.04 (1 H, d, J = 1.2 Hz, S = C-NH-R), 7.99 (1 H, d, J = 8.6 Hz, Ar.), 7.85 (2 H, d, J = 1.4 Hz, Ar.), 7.63 (1 H, d, J = 3.6 Hz, Ar.), 6.86 (1 H, d, J = 3.6 Hz, Ar.), 4.25–4.15 (1 H, m, Cx.), 3.63–3.56 (2 H, m, SO2-CH2), 1.94–1.86 (2 H, m, CH2 propyl), 1.73 (2 H, dt, J = 13.5, 3.6 Hz, Cx.), 1.61 (1 H, dt, J = 12.9, 3.6 Hz, Cx.), 1.56–1.39 (4 H, m, Cx.), 1.29 (2 H, dtd, J = 12.8, 9.3, 3.4 Hz, Cx.), 1.21–1.10 (1 H, m, Cx.), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 175.82 (C═S), 142.64 (C═N), 135.47 (Ar), 130.44 (Ar), 129.54 (Ar), 128.20 (Ar), 123.59 (Ar), 121.26 (Ar), 113.30 (Ar), 108.19 (Ar), 55.19 (Cx.), 52.77 (SO2-CH2), 32.03 (Cx.), 25.32 (Cx.), 25.09 (Cx.), 16.83 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.1 min, 99.98%; ESI-HRMS: [M + H]+: Cal. [M + H]+: 407.15754; Found: 407.15620.
N -(4-nitrophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5 h
Color: yellow; M.P.: 105–107 °C; Yield: 95%; FT-IR (cm− 1): 3269 (N-H), 2969 (Ar. C-H), 1548 (C = N), 1122 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 12.16 (1 H, s, NH-N = C), 10.43 (1 H, s, S = C-NH-R), 8.32 (1 H, s, HC═N), 8.27–8.22 (2 H, m, Ar.), 8.19 (1 H, d, J = 1.5 Hz, Ar.), 8.11–8.06 (2 H, m, Ar.), 7.98 (1 H, dd, J = 8.7, 1.6 Hz, Ar.), 7.89 (1 H, d, J = 8.7 Hz, Ar.), 7.65 (1 H, d, J = 3.6 Hz, Ar.), 6.88 (1 H, dd, J = 3.7, 0.7 Hz, Ar.), 3.64–3.57 (2 H, m, SO2-CH2), 1.51 (2 H, h, J = 7.4 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.5 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 175.35 (C═S), 145.62 (C═N), 144.69 (Ar), 143.63 (Ar), 135.77 (Ar), 130.45 (Ar), 129.10 (Ar), 128.33 (Ar), 124.56 (Ar), 124.40 (Ar), 124.07 (Ar), 123.89 (Ar), 121.92 (Ar), 113.31 (Ar), 108.19 (Ar), 55.22 (SO2-CH2), 16.83 (CH2 propyl), 12.2 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.9 min, 99.91%; ESI-HRMS: [M-H]+ Cal.: 444.08002; Found: 444.08131.
N -(2,4-dimethylphenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5i
Color: white; M.P.: 225 °C; Yield: 91%; FT-IR (cm− 1): 3117 (N-H), 2968 (Ar. C-H), 1502 (C = N), 1200 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.71 (1 H, s, NH-N = C), 9.88 (1 H, s, S = C-NH-R), 8.23 (1 H, s, HC═N), 8.15 (1 H, d, J = 1.6 Hz, Ar.), 7.98–7.93 (1 H, m, Ar.), 7.83 (1 H, d, J = 8.7 Hz, Ar.), 7.62 (1 H, d, J = 3.6 Hz, Ar.), 7.15 (1 H, d, J = 7.9 Hz, Ar.), 7.08 (1 H, d, J = 2.2 Hz, Ar.), 7.04–6.99 (1 H, m, Ar.), 6.83 (1 H, d, J = 3.6 Hz, Ar.), 3.63–3.56 (2 H, m, SO2-CH2), 2.29 (3 H, s, CH3), 2.19 (3 H, s, CH3), 1.50 (2 H, h, J = 7.5 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ:177.06 (C═S), 142.76 (C═N), 135.93 (Ar), 135.71 (Ar), 135.49 (Ar), 135.30 (Ar), 130.76 (Ar), 130.44 (Ar), 129.67 (Ar), 128.69 (Ar), 128.19 (Ar), 126.59 (Ar), 123.84 (Ar), 121.48 (Ar), 113.21 (Ar), 108.17 (Ar), 55.16 (SO2-CH2), 20.77 (CH3), 17.91 (CH3), 16.82 (CH2 propyl), 12.21 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.95 min, 99.95%; ESI-HRMS: [M + H]+ Cal.: 429.14189; Found: 429.14073.
N -(4-bromo-2-fluorophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5j
Color: white; M.P.: 205 °C; Yield: 98%; FT-IR (cm− 1): 3215 (N-H), 2964 (Ar. C-H), 1532 (C = N), 1139 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 12.02 (1 H, s, NH-N = C), 9.96 (1 H, s, S = C-NH-R), 8.26 (1 H, s, HC═N), 8.15 (1 H, d, J = 1.6 Hz, Ar.), 7.94 (1 H, dd, J = 8.7, 1.6 Hz, Ar.), 7.86 (1 H, d, J = 8.7 Hz, Ar.), 7.67–7.60 (2 H, m, Ar.), 7.50 (1 H, t, J = 8.3 Hz, Ar.), 7.44 (1 H, dd, J = 8.6, 2.2 Hz, Ar.), 6.85 (1 H, d, J = 3.6 Hz, Ar.), 3.65–3.57 (2 H, m, SO2-CH2), 1.56–1.46 (2 H, m, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 177.25 (C═S), 158.32 (C═N), 156.65 (Ar), 143.85 (Ar), 135.63 (Ar), 131.97 (Ar), 130.45 (Ar), 129.35 (Ar), 128.29 (Ar), 127.37 (Ar), 127.34 (Ar), 127.19 (Ar), 127.11 (Ar), 123.84 (Ar), 121.65 (Ar), 119.34 (Ar), 119.26 (Ar), 119.19 (Ar), 113.27 (Ar), 108.15 (Ar), 55.19 (SO2-CH2), 16.83 (CH2 propyl), 12.21 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.02 min, 99.76%; ESI-HRMS: [M + H]+ Cal.: 499.01168; Found: 499.00819.
N -(3-methoxyphenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5k
Color: off-white; M.P.: 145 °C; Yield: 96%; FT-IR (cm− 1): 3290 (N-H), 2970 (Ar. C-H), 1535 (C = N), 1217 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.81 (1 H, s, NH-N = C), 10.05 (1 H, s, S = C-NH-R), 8.27 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.6 Hz, Ar.), 7.97 (1 H, dd, J = 8.8, 1.7 Hz, Ar.), 7.86 (1 H, d, J = 8.6 Hz, Ar.), 7.64 (1 H, d, J = 3.6 Hz, Ar.), 7.31 (1 H, t, J = 2.3 Hz, Ar.), 7.27 (1 H, t, J = 8.1 Hz, Ar.), 7.20 (1 H, dd, J = 7.7, 2.0 Hz, Ar.), 6.86 (1 H, d, J = 3.6 Hz, Ar.), 6.78 (1 H, dd, J = 8.2, 2.5 Hz, Ar.), 3.77 (3 H, s, OCH3), 3.65–3.57 (2 H, m, SO2-CH2), 1.51 (2 H, h, J = 7.5 Hz, CH2 propyl), 0.84 (4 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 175.76 (C═S), 159.20 (C═N), 143.44 (Ar), 140.36 (Ar), 135.60 (Ar), 130.44 (Ar), 129.41 (Ar), 128.91 (Ar), 128.24 (Ar), 123.95 (Ar), 121.65 (Ar), 117.90 (Ar), 113.25 (Ar), 111.41 (Ar), 110.89 (Ar), 108.19 (Ar), 55.33 (OCH3), 55.19 (SO2-CH2), 16.83 (CH2 propyl), 12.21 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.89 min, 99.97%; ESI-HRMS: [M + H]+ Cal.: 431.12116; Found: 431.11998.
N -(3,5-bis(trifluoromethyl)phenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5 L
Color: white; M.P.: 205–207 °C; Yield: 96%; FT-IR (cm− 1): 3140 (N-H), 2977 (Ar. C-H), 1573 (C = N), 1204 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 12.19 (1 H, s, NH-N = C), 10.50 (1 H, s, S = C-NH-R), 8.50 (1 H, s, HC═N), 8.32 (1 H, s, Ar.), 8.23–8.14 (1 H, m, Ar.), 8.02–7.94 (1 H, m, Ar.), 7.92–7.85 (2 H, m, Ar.), 7.65 (1 H, dd, J = 8.9, 3.7 Hz, Ar.), 6.88 (2 H, dd, J = 17.8, 3.7 Hz, Ar.), 3.61 (2 H, td, J = 7.8, 3.1 Hz, SO2-CH2), 1.52 (2 H, q, J = 7.5 Hz, CH2 propyl), 0.85 (3 H, td, J = 7.4, 1.9 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 175.77 (C═S), 144.83 (C═N), 141.29 (Ar), 135.78 (Ar), 135.55 (Ar), 130.45 (Ar), 130.28 (Ar), 130.06 (Ar), 129.84 (Ar), 129.47 (Ar), 129.09 (Ar), 128.35 (Ar), 128.26 (Ar), 126.16 (Ar), 125.45 (Ar), 124.35 (Ar), 124.03 (Ar), 123.71 (Ar), 122.54 (Ar), 121.91 (Ar), 121.62 (Ar), 120.73 (Ar), 113.32 (Ar), 108.19 (Ar), 108.16 (Ar), 55.23 (SO2-CH2), 16.83 (CH2 propyl), 12.21 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.14 min, 99.72%; ESI-HRMS: [M + H]+ Cal.: 537.08536; Found: 537.08416.
N-(2,6-dimethylphenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5 m
Color: white; M.P.: 170 °C; Yield: 92%; FT-IR (cm− 1): 3306 (N-H), 2966 (Ar. C-H), 1535 (C = N), 1126 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.71 (1 H, s, NH-N = C), 9.87 (1 H, s, S = C-NH-R), 8.23 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.6 Hz, Ar.), 7.98 (1 H, dd, J = 8.8, 1.7 Hz, Ar.), 7.83 (1 H, d, J = 8.7 Hz, Ar.), 7.62 (1 H, d, J = 3.7 Hz, Ar.), 7.11 (3 H, q, J = 5.2 Hz, Ar.), 6.82 (1 H, d, J = 3.6 Hz, Ar.), 3.63–3.56 (2 H, m, SO2-CH2), 2.20 (6 H, s, CH3), 1.49 (2 H, h, J = 7.5 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 176.88 (C═S), 142.59 (C═N), 137.38 (Ar), 136.65 (Ar), 136.44 (Ar), 135.47 (Ar), 130.43 (Ar), 129.76 (Ar), 128.18 (Ar), 127.72 (Ar), 127.65 (Ar), 127.03 (Ar), 123.91 (Ar), 121.44 (Ar), 113.17 (Ar), 108.17 (Ar), 55.14 (SO2-CH2), 18.20 (CH3), 16.82 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.91 min, 99.94%; ESI-HRMS: [M + H]+ Cal.: 429.14189; Found: 429.14087.
2-([1-(propylsulfonyl)−1 H-indol-5-yl]methylene)-N-(2-(trifluoromethyl)phenyl)hydrazinecarbothioamide 5n
Color: off-white; M.P.: 215–217 °C; Yield: 91%; FT-IR (cm− 1): 3330 (N-H), 2972 (Ar. C-H), 1540 (C = N), 1211 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 12.01 (1 H, s, NH-N = C), 9.99 (1 H, s, S = C-NH-R), 8.27 (1 H, s, HC═N), 8.11 (1 H, d, J = 1.6 Hz, Ar.), 7.92 (1 H, dd, J = 8.8, 1.7 Hz, Ar.), 7.86 (1 H, d, J = 8.7 Hz, Ar.), 7.77 (1 H, d, J = 7.9 Hz, Ar.), 7.74–7.68 (2 H, m, Ar.), 7.64 (1 H, d, J = 3.7 Hz, Ar.), 7.51 (1 H, dt, J = 8.3, 4.3 Hz, Ar.), 6.85 (1 H, dd, J = 3.7, 0.8 Hz, Ar.), 3.65–3.56 (2 H, m, SO2-CH2), 1.57–1.46 (2 H, m, CH2 propyl), 0.85 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 177.36 (C═S), 143.57 (C═N), 137.52 (Ar), 135.58 (Ar), 132.66 (Ar), 131.81 (Ar), 130.45 (Ar), 129.34 (Ar), 128.25 (Ar), 127.27 (Ar), 126.31 (Ar), 126.27 (Ar), 126.23 (Ar), 126.00 (Ar), 125.77 (Ar), 124.90 (Ar), 123.45 (Ar), 121.70 (Ar), 113.36 (Ar), 108.13 (Ar), 55.15 (SO2-CH2), 16.81 (CH2 propyl), 12.18 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.94 min, 99.75%; ESI-HRMS: [M + H]+ Cal.: 469.09798; Found: 469.09709.
N -(3-fluorophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5o
Color: off-white; M.P.: 169 °C; Yield: 94%; FT-IR (cm− 1): 3291 (N-H), 2971 (Ar. C-H), 1560 (C = N), 1142 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.92 (1 H, s, NH-N = C), 10.16 (1 H, s, S = C-NH-R), 8.28 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.6 Hz, Ar.), 7.97 (1 H, dd, J = 8.7, 1.6 Hz, Ar.), 7.87 (1 H, d, J = 8.7 Hz, Ar.), 7.64 (2 H, tt, J = 4.6, 2.3 Hz, Ar.), 7.51–7.46 (1 H, m, Ar.), 7.40 (1 H, td, J = 8.2, 6.7 Hz, Ar.), 7.07–6.98 (1 H, m, Ar.), 6.87 (1 H, dd, J = 3.5, 0.7 Hz, Ar.), 3.69–3.55 (2 H, m, SO2-CH2), 1.60–1.38 (2 H, m, CH2 propyl), 0.85 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 175.73 (C═S), 162.62 (C═N), 160.70 (Ar), 143.84 (Ar), 141.04 (Ar), 140.95 (Ar), 135.64 (Ar), 130.42 (Ar), 129.66 (Ar), 129.58 (Ar), 129.29 (Ar), 128.24 (Ar), 123.97 (Ar), 121.70 (Ar), 121.38 (Ar), 121.36 (Ar), 113.24 (Ar), 112.39 (Ar), 112.19 (Ar), 111.92 (Ar), 111.75 (Ar), 108.16 (Ar), 55.20 (SO2-CH2), 16.82 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.91 min, 99.97%; ESI-HRMS: [M + H]+ Calculated: 419.10117; Found: 419.10006. HSQC Correlations: 13C NMR-1H NMR (126–500 MHz, DMSO) δ 144.13–8.29, 122.05–8.19, 124.31–7.99, 113.51–7.88, 128.50–7.66, 112.87–7.65, 121.73–7.49, 129.95–7.42, 112.22–7.05, 108.52–6.88, 55.50–3.63, 40.35–2.50, 17.15–1.53, 12.48–0.86.
N -(4-methoxyphenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5p
Color: white; M.P.: 142–144 °C; Yield: 96%; FT-IR (cm− 1): 3228 (N-H), 2970 (Ar. C-H), 1531 (C = N), 1140 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.71 (1 H, s, NH-N = C), 9.99 (1 H, s, S = C-NH-R), 8.25 (1 H, s, HC═N), 8.16 (1 H, d, J = 1.7 Hz, Ar.), 7.96 (1 H, dd, J = 8.7, 1.7 Hz, Ar.), 7.85 (1 H, d, J = 8.7 Hz, Ar.), 7.63 (1 H, d, J = 3.6 Hz, Ar.), 7.45–7.38 (2 H, m, Ar.), 6.96–6.90 (2 H, m, Ar.), 6.85 (1 H, d, J = 3.6 Hz, Ar.), 3.77 (3 H, s, OCH3), 3.64–3.57 (2 H, m, SO2-CH2), 1.51 (2 H, h, J = 7.4 Hz, CH2 propyl), 0.85 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 176.42 (C═S), 157.10 (C═N), 143.04 (Ar), 135.52 (Ar), 132.16 (Ar), 130.42 (Ar), 129.53 (Ar), 128.19 (Ar), 127.69 (Ar), 123.89 (Ar), 121.50 (Ar), 113.42 (Ar), 113.19 (Ar), 108.16 (Ar), 55.40 (OCH3), 55.18 (SO2-CH2), 16.82 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.84 min, 99.87%; ESI-HRMS: [M + H]+: Cal. 431.12116; Found: 431.11994.
N -(4-chlorophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5q
Color: yellow; M.P.:152–154 °C; Yield: 97%; FT-IR (cm− 1): 3300 (N-H), 2969 (Ar. C-H), 1545 (C = N), 1196 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.87 (1 H, s, NH-N = C), 10.14 (1 H, s, S = C-NH-R), 8.27 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.5 Hz, Ar.), 7.97 (1 H, dd, J = 8.7, 1.6 Hz, Ar.), 7.86 (1 H, d, J = 8.7 Hz, Ar.), 7.64 (3 H, dd, J = 6.3, 2.5 Hz, Ar.), 7.46–7.39 (2 H, m, Ar.), 6.86 (1 H, dd, J = 3.7, 0.8 Hz, Ar.), 3.70–3.49 (2 H, m, SO2-CH2), 1.60–1.42 (2 H, m, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ:176.01 (C═S), 143.70 (C═N), 138.26 (Ar), 135.61 (Ar), 130.42 (Ar), 129.41 (Ar), 129.35 (Ar), 128.24 (Ar), 128.08 (Ar), 127.57 (Ar), 123.95 (Ar), 121.65 (Ar), 113.22 (Ar), 108.16 (Ar), 55.20 (SO2-CH2), 16.82 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.01 min, 99.82%; ESI-HRMS: [M-H]+ Cal.: 433.05597; Found: 433.05717.
N -(3-(methylthio)propyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5r
Color: off-white; M.P.: 106 °C; Yield: 93%; FT-IR (cm− 1): 3324 (N-H), 2960 (Ar. C-H), 1513 (C = N), 1140 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.42 (1 H, s, NH-N = C), 8.57 (1 H, t, J = 6.0 Hz), 8.16 (1 H, s, S = C-NH-R), 8.05 (1 H, s, HC═N), 7.90–7.81 (2 H, m, Ar.), 7.63 (1 H, d, J = 3.7 Hz, Ar.), 6.85 (1 H, d, J = 3.7 Hz, Ar.), 3.65 (2 H, q, J = 6.7 Hz, CH2, Ar.), 3.62–3.55 (2 H, m, SO2-CH2), 2.55–2.46 (2 H, m, CH2), 2.06 (3 H, s, S-CH3), 1.88 (2 H, p, J = 7.2 Hz, CH2), 1.51 (2 H, h, J = 7.5 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 177.15 (C═S), 142.41 (C═N), 135.41 (Ar), 130.42 (Ar), 129.67 (Ar), 128.19 (Ar), 123.49 (Ar), 121.18 (Ar), 113.23 (Ar), 108.13 (Ar), 55.17 (SO2-CH2), 42.89 (aliph.), 30.93 (aliph.), 28.51 (aliph.), 16.81 (aliph.), 14.81 (CH2 propyl), 12.20 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.91 min, 99.98%; ESI-HRMS: [M + H]+ Cal.: 413.11396; Found: 413.11303.
N -(2,6-dichlorophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5s
Color: off-white; M.P.: 212 °C; Yield: 96%; FT-IR (cm− 1): 3328 (N-H), 2974 (Ar. C-H), 1541 (C = N), 1123 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.96 (1 H, s, NH-N = C), 10.09 (1 H, s, S = C-NH-R), 8.25 (1 H, s, HC═N), 8.20–8.16 (1 H, m, Ar), 7.97 (1 H, dd, J = 8.8, 1.6 Hz, Ar), 7.85 (1 H, d, J = 8.8 Hz, Ar), 7.64 (1 H, d, J = 3.7 Hz, Ar), 7.55 (2 H, d, J = 8.1 Hz, Ar), 7.38 (1 H, t, J = 8.1 Hz, Ar), 6.85 (1 H, dd, J = 3.7, 0.8 Hz, Ar), 3.70–3.51 (2 H, m, SO2-CH2), 1.58–1.44 (2 H, m, CH2 propyl), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 177.30 (C═S), 143.38 (C═N), 135.56 (Ar), 135.39 (Ar), 135.32 (Ar), 130.41 (Ar), 129.47 (Ar), 128.44 (Ar), 128.23 (Ar), 123.92 (Ar), 121.55 (Ar), 113.20 (Ar), 108.14 (Ar), 55.17 (SO2-CH2), 16.81 (CH2 propyl), 12.19 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.81 min, 99.95%; ESI-HRMS: [M + H]+ Cal.: 469.03265; Found: 469.03189.
N -(2,6-difluorophenyl)−2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)hydrazinecarbothioamide 5t
Color: off-white; M.P.: 200–202 °C; Yield: 92%; FT-IR (cm− 1): 3335 (N-H), 2977 (Ar. C-H), 1547 (C = N), 1148 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 12.06 (1 H, s, NH-N = C), 9.81 (1 H, s, S = C-NH-R), 8.26 (1 H, s, HC═N), 8.17 (1 H, d, J = 1.7 Hz, Ar), 7.96 (1 H, dd, J = 8.8, 1.6 Hz, Ar), 7.86 (1 H, d, J = 8.7 Hz, Ar), 7.64 (1 H, d, J = 3.6 Hz, Ar), 7.45–7.39 (1 H, m, Ar), 7.18 (2 H, t, J = 8.0 Hz, Ar), 6.85 (1 H, dd, J = 3.7, 0.8 Hz, Ar), 3.70–3.53 (2 H, m, SO2-CH2), 1.62–1.46 (2 H, m, CH2 propyl), 0.84 (3 H, t, J = 7.5 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 178.13 (C═S), 160.21 (C═N), 160.18 (Ar), 158.24 (Ar), 158.20 (Ar), 143.86 (Ar), 135.61 (Ar), 130.42 (Ar), 129.38 (Ar), 129.17 (Ar), 129.09 (Ar), 128.26 (Ar), 123.89 (Ar), 121.60 (Ar), 117.18 (Ar), 117.05 (Ar), 116.92 (Ar), 113.22 (Ar), 111.94 (Ar), 111.90 (Ar), 111.75 (Ar), 108.13 (Ar), 55.19 (SO2-CH2), 16.82 (CH2 propyl), 12.19 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 2.91 min, 99.98%; ESI-HRMS: [M + H]+ Cal.: 437.09175; Found: 437.09061.
N -isobutyl-2-([1-(propylsulfonyl)−1 H -indol-5-yl]methylene)shydrazinecarbothioamide 5u
Color: white; M.P.: 155–157 °C; Yield: 95%; FT-IR (cm− 1): 3369 (N-H), 2954 (Ar. C-H), 1540 (C = N), 1140 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 8.47 (1 H, t, J = 6.0 Hz, S = C-NH-R), 8.17 (1 H, s, HC═N), 8.04 (1 H, t, J = 1.1 Hz, NH-N = C), 7.90–7.82 (2 H, m, Ar), 7.63 (2 H, d, J = 3.6 Hz, Ar), 6.86 (1 H, d, J = 3.6 Hz, Ar), 3.64–3.56 (2 H, m, SO2-CH2), 3.46–3.37 (2 H, m, N-CH2), 2.03 (1 H, dt, J = 13.6, 6.8 Hz, CH), 1.55–1.46 (2 H, m, CH2 propyl), 0.89 (6 H, d, J = 6.7 Hz, CH3), 0.84 (3 H, t, J = 7.4 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ:177.35 (C═S), 142.36 (C═N), 135.40 (Ar), 130.42 (Ar), 129.67 (Ar), 128.17 (Ar), 123.43 (Ar), 121.20 (Ar), 113.27 (Ar), 108.14 (Ar), 55.16 (SO2-CH2), 50.94 (iso but.), 28.01 (iso but.), 20.24 (iso but.), 16.81 (CH2 propyl), 12.19 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.02 min, 99.87%; ESI-HRMS: [M + H]+; Cal.: 381.14189; Found: 381.14093.
2-([1-(propylsulfonyl)−1H-indol-5-yl]methylene)-N-tritylhydrazinecarbothioamide 5v
Color: white; M.P.: 236 °C; Yield: 95%; FT-IR (cm− 1): 3306 (N-H), 2972 (Ar. C-H), 1518 (C = N), 1157 (C = S); 1H-NMR (500 MHz, DMSO-d6) δ: 11.75 (1 H, s, NH-N = C), 9.11 (1 H, s, S = C-NH-R), 8.26 (1 H, s, HC═N), 8.01–7.81 (2 H, m, Ar.), 7.68 (2 H, d, J = 11.1 Hz, Ar.), 7.33 (15 H, dq, J = 20.4, 10.8, 9.2 Hz, Ar.), 6.88 (1 H, s, Ar.), 3.62 (2 H, t, J = 7.5 Hz, SO2-CH2), 1.51 (2 H, q, J = 7.6 Hz, CH2 propyl), 0.84 (3 H, t, J = 7.5 Hz, CH3 propyl); 13C-NMR (125 MHz, DMSO-d6) δ: 177.20 (C═S), 144.94 (C═N), 142.38 (Ar), 135.80 (Ar), 130.89 (Ar), 129.58 (Ar), 129.18 (Ar), 128.71 (Ar), 128.24 (Ar), 127.30 (Ar), 122.75 (Ar), 121.74 (Ar), 113.99 (Ar), 108.49 (Ar), 71.64 (CH3), 55.46 (SO2-CH2), 17.14 (CH2 propyl), 12.52 (CH3 propyl); HPLC-PDA: MeCN/MeOH (1/1), λ = 254 nm, Rt: 3.29 min, 99.89%; ESI-HRMS:[M-H]+ Cal.: 565.17319; Found: 565.17515.
Biological studies
Enzyme preparation
The enzymes were obtained by transfection of COS-7 cells using plasmids encoding human (h-NTPDase1 (GenBank accession no. U87967)59, h-NTPDase2 (GenBank accession no. NM_203468)60 a kind gift of Aileen F. Knowles, h-NTPDase3 (GenBank accession no. AF034840)61 a kind gift of Terence L. Kirley, h-NTPDase8 (GenBank accession no. AY430414)62 and h-e5’NT (GenBank accession no. DQ186653)63, as previously described64. Briefly, COS-7 cells were transfected in a 10 cm dish plate by incubating them for 5 h at 37 °C in a serum-free DMEM/F-12 containing 6 µg plasmid DNA sample and 24 µL Lipofectamine reagent. Following this incubation, an equal volume of DMEM/F-12 containing 20% fetal bovine serum was added to the plate. After an incubation period of 44–48 h, cells were washed 3 times with a harvesting buffer containing 95 mM NaCl, 0.1 mM PMSF, and 45 mM Tris at pH 7.5. The cells were then scraped, centrifuged twice (300 g, 5 min. each time, at 4 °C), and resuspended in the harvesting buffer supplemented with aprotinin (10 µg/mL) before being sonicated. To separate the nuclear and cell debris, centrifugation at 850 g for 5 min. at 4 °C was performed. The obtained supernatant was mixed with 7.5% glycerol and stored at −80 °C. The protein concentration was measured using the Bradford microplate test, with bovine serum albumin as the standard reference protein.
NTPDase enzyme activity assays
The synthesized compounds were tested against h-NTPDase1, −2, −3, and − 8 using a colorimetric technique with malachite green dye as per the previously described method and minor changes. Compounds were prepared in 10% DMSO and evaluated at 100 µM. Optimized enzyme concentrations were h-NTPDase1 (12 ng/well), h-NTPDase2 (37 ng/well), h-NTPDase3 (43 ng/well), and h-NTPDase8 (63 ng/well), using a Tris-HCl buffer (10 mM Tris-Base, 1 mM CaCl2, 2 mM MgCl2, pH 7.4). The assay began by adding 10 µL of the test compound with 55 µL of assay buffer and 10 µL of enzyme solution in a 96-well plate, incubating for 10 min at 37 C. ATP (100 µM) was added as the substrate to start the reaction, which was stopped after 15 min by adding malachite green reagent. Absorbance was measured at 630 nm. Compounds with over 50% inhibition of any isoform were further tested to determine IC50 values using GraphPad Prism 5.016.
CD73 enzyme activity assay
The e5′NT inhibition assays were conducted on human enzymes using established protocols65. Test substances were prepared to achieve a final concentration of 100 mM in a 10% DMSO solution. AMP, at a concentration of 2 mM, was used as the substrate to initiate enzymatic activity during incubation. The enzyme concentration was optimized to 40 ng/well in a Tris buffer solution comprising 10 mM Tris-HCl, 5 mM CaCl2, and 5 mM MgCl2, with the pH adjusted to 7.4 for optimal conditions. To ensure accuracy and reproducibility, all assays were performed in triplicate. The process began with the addition of 55 µL of assay buffer to each well, followed by 10 µL of the e5′NT enzyme solution. Then, 10 µL of the test compound solution was introduced to obtain a final reaction volume of 100 µL per well. The mixture was incubated at 37 °C for 10 min. Subsequently, the reaction was initiated by adding 10 µL of AMP (2 mM) as the substrate. The reaction proceeded for 15 min at 37 °C to allow sufficient enzyme-substrate interaction16,17,66.
The release of phosphate, resulting from the enzymatic hydrolysis of AMP, was measured using 15 µL of malachite green reagent. Absorbance changes were recorded at 630 nm to quantify the phosphate levels. Test compounds exhibiting inhibition levels greater than 50% were further analyzed through serial dilutions, ranging from 7 to 8 steps, to determine their IC50 values. Nonlinear regression analysis was performed using GraphPad Prism® 5.0 software (San Diego, CA, USA) to accurately calculate IC50 values, providing insight into the inhibitory potential of each compound67.
Computational studies
All computational studies were performed using the Schrödinger Maestro suite (Maestro, version 2025-1; Schrödinger, LLC, New York, NY, USA). Molecular docking calculations were carried out using Glide, while molecular dynamics (MD) simulations were performed with the Desmond (Maestro version 2025-1; D. E. Shaw Research)68 module implemented in Maestro. The crystal structure of human ecto-5′-nucleotidase (e-5′NT, CD73) (PDB ID: 7PBY; resolution: 1.13 Å) was retrieved from the Protein Data Bank67. The protein was prepared using the Protein Preparation Wizard, including bond order assignment, addition of missing hydrogen atoms, optimization of protonation states at physiological pH, and restrained minimization using the OPLS4 force field57.
The co-crystallized ligand (4-nitrocatechol) was used to define the active site, and the receptor grid was generated accordingly. The Glide grid box was centered on the co-crystallized ligand with a grid box size of approximately 20 × 20 × 20 Å, ensuring full coverage of the active site. Ligands were prepared using LigPrep to generate optimized 3D structures and appropriate ionization states at physiological pH69.
Docking calculations were performed using the Glide XP (extra precision) protocol. To account for receptor flexibility, Induced Fit Docking (IFD) was carried out with an initial softened-potential Glide docking step, followed by Prime refinement of residues within 5.0 Å of the ligand. The IFD grid box was defined with an inner box of 10 × 10 × 10 Å and an outer box of 25 × 25 × 25 Å centered on the active site. Final redocking was performed using Glide XP, and the best-ranked poses were selected based on docking scores and interaction profiles70.
The selected ligand–protein complexes were subjected to 500 ns MD simulations under NPT conditions (300 K, 1 atm) using the OPLS4 force field and the TIP4P water model. Systems were solvated in an orthorhombic box with a buffer distance of 10 Å and neutralized with appropriate counterions. Trajectory analyses included root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (rGyr), solvent-accessible surface area (SASA), and molecular surface area (MolSA). In addition, protein–ligand interaction profiles and fractional interaction histograms were evaluated to identify key residues contributing to binding throughout the simulation71,72,73. Furthermore, in silico ADME properties were predicted using the QikProp module to assess drug-likeness and pharmacokinetic behavior74.
Conclusion
In this study, a library of N-propylsulfonyl-substituted indole-based thiosemicarbazones (5a–5v) was successfully synthesized and very well characterized using FT-IR, NMR, HRMS, and HPLC analyses.
The synthesized compounds were evaluated against e-5′NT and NTPDase1, −2, −3, and − 8, where several derivatives exhibited potent inhibitory activity in the low micromolar range. Notably, compounds 5n (IC50 = 1.7 µM, e-5′NT), 5i (IC50 = 1.6 µM, NTPDase1), 5f (IC50 = 1.1 µM, NTPDase2; 1.0 µM, NTPDase8), and 5o (IC50 = 1.7 µM, NTPDase3; 1.1 µM, NTPDase8) emerged as the most active inhibitors. In addition, several compounds displayed notable isoform selectivity, indicating the influence of substituent type and position on enzyme specificity.
Molecular docking studies against e-5′NT (PDB ID: 7PBY) revealed that the active compounds adopt similar binding orientations within the catalytic pocket, with key interactions involving residues such as Asp-506, Phe-500, Phe-417, Asn-390, and Arg-395. Docking scores were in a narrow range (− 7.200 to − 7.839 kcal/mol), while MM-GBSA analysis provided better discrimination, highlighting 5n (− 71.94 kcal/mol), 5 g (− 71.21 kcal/mol), and 5o (− 71.05 kcal/mol) as the most stable binders. Energy decomposition analysis further demonstrated that van der Waals and lipophilic interactions are the dominant contributors to binding.
Molecular dynamics simulations (500 ns) of the 5n– and 5o–e-5′NT complexes confirmed the stability of the binding interactions. Both systems showed low RMSD values (protein: ~1.2–1.4 Å; ligand: ~2.4–3.0 Å) and minimal RMSF fluctuations (~ 0.9–1.25 Å). Persistent interactions with key residues, particularly Asp-506 and Phe-500 (up to ~ 95–100% occupancy), along with stable rGyr (~ 5.8–6.4 Å), SASA, MolSA, and PSA profiles, indicate compact ligand behavior and consistent binding throughout the simulation. In silico ADME analysis revealed that the majority of compounds possess favorable pharmacokinetic properties, including high oral absorption and good membrane permeability. Among them, 5n and 5o exhibited well-balanced ADME profiles, further supporting their potential as drug-like candidates.
Overall, the integration of synthetic, biological, and computational results demonstrates that the indole-based thiosemicarbazone scaffold represents a promising framework for the development of potent and selective ectonucleotidase inhibitors, with compounds 5n and 5o identified as the most promising leads for further optimization.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files.
References
Grković, I., Drakulić, D., Martinović, J. & Mitrović, N. Role of ectonucleotidases in synapse formation during brain development: physiological and pathological implications. Curr. Neuropharmacol. 17 (1), 84–98 (2019).
Lee, S-Y. & Müller, C. E. Nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) and its inhibitors. MedChemComm 8 (5), 823–840 (2017).
Nabinger, D. D., Altenhofen, S. & Bonan, C. D. Zebrafish models: Gaining insight into purinergic signaling and neurological disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry. 98, 109770 (2020).
Caiazzo, E., Bilancia, R., Rossi, A., Ialenti, A. & Cicala, C. Ectonucleoside triphosphate diphosphohydrolase-1/CD39 affects the response to ADP of female rat platelets. Front. Pharmacol. 10, 1689 (2020).
Baqi, Y. Ecto-nucleotidase inhibitors: recent developments in drug discovery. Mini Rev. Med. Chem. 15 (1), 21–33 (2015).
Hechler, B. & Gachet, C. Purinergic receptors in thrombosis and inflammation. Arterioscler. Thromb. Vasc. Biol. 35 (11), 2307–2315 (2015).
Bagatini, M. D. et al. The impact of purinergic system enzymes on noncommunicable, neurological, and degenerative diseases. J. Immunol. Res. 2018 (1), 4892473 (2018).
Saunders, D. C. et al. Ectonucleoside triphosphate diphosphohydrolase-3 antibody targets adult human pancreatic β cells for in vitro and in vivo analysis. Cell Metabol. 29 (3), 745–754 (2019). e4.
Zhong, A. H., Gordon Jiang, Z., Cummings, R. D. & Robson, S. C. Various N-glycoforms differentially upregulate E-NTPDase activity of the NTPDase3/CD39L3 ecto-enzymatic domain. Purinergic Signal. 13, 601–609 (2017).
Fausther, M. et al. Coexpression of ecto-5′-nucleotidase/CD73 with specific NTPDases differentially regulates adenosine formation in the rat liver. Am. J. Physiology-Gastrointestinal Liver Physiol. 302 (4), G447–G59 (2012).
Zimmermann, H., Zebisch, M. & Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 8, 437–502 (2012).
Longhi, M. S., Robson, S. C., Bernstein, S. H., Serra, S. & Deaglio, S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J. Mol. Med. 91, 165–172 (2013).
Murphy, P. S. et al. CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell. Death Differ. 24 (3), 559–570 (2017).
Bowser, J. L. & Broaddus, R. R. CD73s protection of epithelial integrity: thinking beyond the barrier. Tissue Barriers. 4 (4), 220–238 (2016).
Sadej, R. & Skladanowski, A. Dual, enzymatic and non-enzymatic, function of ecto-5’-nucleotidase (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochim. Pol. 59 (4), 647–652 (2012).
Aftab, H. et al. Synthesis, Biological Evaluation and Molecular Docking Studies of Novel 4-Propylsulfonylpiperazines‐Based Thiosemicarbazones as Ecto‐5′‐Nucleotidase and NTPDase Inhibitors. Arch. Pharm. 358 (9), e70098 (2025).
Batool, Z. et al. N-hexylsulfonyl indole based thiosemicarbazones as potent and selective ecto-5′-nucleotidase and NTPDase inhibitors. Bioorg. Chem. 163, 108717 (2025).
Jadoon, M. S. K., Pelletier, J., Sévigny, J. & Iqbal, J. Synthesis of new class of indole acetic acid sulfonate derivatives as ectonucleotidases inhibitors. RSC Adv. 13 (42), 29496–29511 (2023).
Murtaza, A. et al. Divergent synthesis and elaboration of structure activity relationship for quinoline derivatives as highly selective NTPDase inhibitor. Bioorg. Chem. 115, 105240 (2021).
Willig, J. B. et al. Imatinib mesylate affects extracellular ATP catabolism and expression of NTPDases in a chronic myeloid leukemia cell line. Purinergic Signal. 16, 29–40 (2020).
Ateşoğlu, Ş. et al. Synthesis, Characterization and In Vitro and In Silico Biological Evaluation of New Mannich-Based Rhodanine and Thiazolidine-2, 4-dione Derivatives as Potential Anti-Lung-Cancer Agents. Synlett. (2025).
Batool, Z. et al. Design, synthesis, and in vitro and in silico study of 1-benzyl-indole hybrid thiosemicarbazones as competitive tyrosinase inhibitors. RSC Adv. 14 (39), 28524–28542 (2024).
Ghaffar, U. et al. Synthesis, Anti-Alzheimer Evaluation and In Silico Study of 4‐Methoxyphenyl) Sulfonyl Indole Hybrid Thiosemicarbazones. Arch. Pharm. 358 (7), e70034 (2025).
Russo, E. et al. Indole antitumor agents in nanotechnology formulations: an overview. Pharmaceutics 15 (7), 1815 (2023).
Hawash, M. et al. Novel indole-pyrazole hybrids as potential tubulin-targeting agents; Synthesis, antiproliferative evaluation, and molecular modeling studies. J. Mol. Struct. 1285, 135477 (2023).
Mohamed, M. R., Shoman, M. E., Ali, T. F. & Abuo-Rahma, G. E. D. A. Anti-tumor Activity of Indole: A Review. Lett. Drug Des. Discovery. 21 (16), 3332–3348 (2024).
Anuradha, M. A. & Sharma, S. Indole as an emerging scaffold in anticancer drug design. AIP Conference Proceedings: AIP Publishing LLC; p. 020198. (2023).
Rana, M., Ranjan, R., Sekhar Ghosh, N., Kumar, D. & Singh, R. A Review on Indole as a Cardinal Scaffold for Anticancer Drugs Development. Curr. Cancer Therapy Reviews. 20 (4), 372–385 (2024).
Mehra, A. et al. Indole derived anticancer agents. ChemistrySelect 7 (34), e202202361 (2022).
Batool, Z. et al. Dual targeting of neuroblastoma and cholinesterase by morpholino/pyrrolidino-sulfonyl-indole thiosemicarbazones: Synthesis, characterization, enzyme inhibition, cytotoxicity, docking and dynamics studies. Bioorg. Chem. 167, 109252. (2025).
Tokali, F. S., Şenol, H., Ateşoğlu, Ş., Tokalı, P. & Akbaş, F. Design, synthesis, and cytotoxic evaluation of new thiosemicarbazone/thiazolidin-4-one derivatives on PC3 cells. Future Med. Chem. 1–12. https://doi.org/10.1080/17568919.2025.2592533 (2025).
Garbuz, O. et al. Thiosemicarbazone-Based Compounds: Cancer Cell Inhibitors with Antioxidant Properties. Molecules 30 (9), 2077 (2025).
Kozyra, P. et al. Potential anticancer agents against melanoma cells based on an as-synthesized thiosemicarbazide derivative. Biomolecules 12 (2), 151 (2022).
Balakrishnan, N. et al. Influence of Indole-N substitution of thiosemicarbazones in cationic Ru (II)(η6-Benzene) complexes on their anticancer activity. Organometallics 42 (3), 259–275 (2023).
Sibuh, B. Z. et al. Synthesis, in silico study, and anti-cancer activity of thiosemicarbazone derivatives. Biomedicines 9 (10), 1375 (2021).
PapeVeronika, F., EnyedyÉva, A., KepplerBernhard, K. & KowolChristian, R. Anticancer thiosemicarbazones: chemical properties, interaction with iron metabolism, and resistance development (Antioxidants & redox signaling, 2019).
Singh, N. K., Kumbhar, A. A., Pokharel, Y. R. & Yadav, P. N. Anticancer potency of copper (II) complexes of thiosemicarbazones. J. Inorg. Biochem. 210, 111134 (2020).
Sardroud, S. J., Hosseini-Yazdi, S. A., Mahdavi, M., Poupon, M. & Skorepova, E. Synthesis, characterization and in vitro evaluation of anticancer activity of a new water-soluble thiosemicarbazone ligand and its complexes. Polyhedron 175, 114218 (2020).
Manakkadan, V. et al. Synthesis and characterization of N4-substituted thiosemicarbazones: DNA/BSA binding, molecular docking, anticancer activity, ADME study and computational investigations. J. Mol. Struct. 1285, 135494 (2023).
Haribabu, J. et al. Michael addition-driven synthesis of cytotoxic palladium (ii) complexes from chromone thiosemicarbazones: investigation of anticancer activity through in vitro and in vivo studies. New J. Chem. 47 (33), 15748–15759 (2023).
Preetha, K., Seena, E., Maniyampara, P. K., Manoj, E. & Kurup, M. P. Synthesis, crystal structure, Hirshfeld surface analysis, DFT, molecular docking and in vitro antitumor studies of (2E)-2-[4-(diethylamino) benzylidene]-N-ethylhydrazinecarbothioamide. J. Mol. Struct. 1295, 136700 (2024).
Tokalı, F. S., Şenol, H., Katmerlikaya, T. G., Dağ, A. & Şendil, K. Novel thiosemicarbazone and thiazolidin-4‐one derivatives containing vanillin core: Synthesis, characterization, and anticancer activity studies. J. Heterocycl. Chem. 60 (4), 645–656. https://doi.org/10.1002/jhet.4619 (2023).
Tokalı, F. S., Taslimi, P., Usanmaz, H., Karaman, M. & Sendil, K. Synthesis, characterization, biological activity and molecular docking studies of novel schiff bases derived from thiosemicarbazide: Biochemical and computational approach. J. Mol. Struct. 1231 https://doi.org/10.1016/j.molstruc.2020.129666 (2021).
Şenol, H. et al. 2-Propyl‐3‐Aminoquinazoline‐4 (3H)‐one Derivatives as Potential Androgen Receptor Inhibitors: Synthesis and Cytotoxicity Against PC3 Cell Line via In Vitro and In Silico Studies. Arch. Pharm. 358 (7), e70048 (2025).
Afzal, S. et al. Functionalized oxoindolin hydrazine carbothioamide derivatives as highly potent inhibitors of nucleoside triphosphate diphosphohydrolases. Front. Pharmacol. 11, 585876 (2020).
Zaigham, Z. H. et al. Synthesis and biological evaluation of sulfamoyl benzamide derivatives as selective inhibitors for h-NTPDases. RSC Adv. 13 (30), 20909–20915 (2023).
Khan, K. M. et al. Schiff bases of tryptamine as potent inhibitors of nucleoside triphosphate diphosphohydrolases (NTPDases): structure-activity relationship. Bioorg. Chem. 82, 253–266 (2019).
Tasleem, M. et al. Synthesis, in vitro, and in silico studies of morpholine-based thiosemicarbazones as ectonucleotide pyrophosphatase/phosphodiesterase-1 and-3 inhibitors. Int. J. Biol. Macromol. 266, 131068 (2024).
Zou, Y. et al. New triazole derivatives as antifungal agents: Synthesis via click reaction, in vitro evaluation and molecular docking studies. Bioorg. Med. Chem. Lett. 22 (8), 2959–2962 (2012).
Çakır, F. et al. Novel 4-((3-fluorobenzyl)oxy)benzohydrazide derivatives as promising anti-prostate cancer agents: Synthesis, characterization and in vitro & in silico biological activity studies. J. Mol. Struct. 1322, 140702. https://doi.org/10.1016/j.molstruc.2024.140702 (2025).
Zengin Kurt, B., Özmen, Ö., Öztürk Civelek, D., Şenol, H. & Sonmez, F. Exploring anticancer properties of new triazole-linked benzenesulfonamide derivatives against colorectal carcinoma: Synthesis, cytotoxicity, and in silico insights. Bioorg. Med. Chem. 119, 118060. https://doi.org/10.1016/j.bmc.2025.118060 (2025).
Raza, S. et al. Targeting Lung Cancer: Synthesis, Characterization, Enzyme Inhibition, and Antiproliferative Activity of 1H-Benzo[f]chromen-1-One-Thiosemicarbazones Supported by Molecular Docking. Arch. Pharm. 358 (10), e70116. https://doi.org/10.1002/ardp.70116 (2025).
Şenol, H. & Çakır, F. 3-Amino-thiophene-2-carbohydrazide Derivatives as Anti Colon Cancer Agents: Synthesis, Characterization, In-Silico and In-Vitro. Biol. Activity Stud. ChemistrySelect. 8 (39), e202302448. https://doi.org/10.1002/slct.202302448 (2023).
Halder, D., Banerjee, S., Biswas, S., Adhikari, N. & Ghosh, B. Molecular modeling to simulation: insights into Gaussian QSAR, molecular docking, and DFT for identification of HDAC3 inhibitors for neurocognitive vascular dementia. Sci. Rep. 15 https://doi.org/10.1038/s41598-025-28304-y (2025).
Al-Khafaji, K. & Taskin Tok, T. Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput. Methods Programs Biomed. 195, 105660. https://doi.org/10.1016/j.cmpb.2020.105660 (2020).
Lanka, G., Banerjee, S., Adhikari, N. & Ghosh, B. Fragment-based discovery of new potential DNMT1 inhibitors integrating multiple pharmacophore modeling, 3D-QSAR, virtual screening, molecular docking, ADME, and molecular dynamics simulation approaches. Mol. Divers. 29 (1), 117–137. https://doi.org/10.1007/s11030-024-10837-5 (2025).
Tokalı, P. et al. New quinazolinone-thiazolidinedione hybrids as selective anti-lung cancer agents and promising EGFR inhibitors. Future Med. Chem. 18 (4), 415–428. https://doi.org/10.1080/17568919.2026.2620368 (2026).
Tokalı, F. S. et al. Targeting cholinergic dysfunction and neuroinflammation through rationally designed Thieno[3,2-d]pyrimidine hybrids. Bioorg. Chem. 175, 109778. https://doi.org/10.1016/j.bioorg.2026.109778 (2026).
Kaczmarek, E. et al. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 271 (51), 33116–33122 (1996).
Knowles, A. F. & Chiang, W-C. Enzymatic and transcriptional regulation of human ecto-ATPase/E-NTPDase 2. Arch. Biochem. Biophys. 418 (2), 217–227 (2003).
Smith, T. M. & Kirley, T. L. Cloning, sequencing, and expression of a human brain ecto-apyrase related to both the ecto-ATPases and CD39 ecto-apyrases1. Biochim Biophys Acta. 1386(1), 65–78. (1998). doi: 10.1016/s0167-4838(98)00063-6
Fausther, M. et al. Cloning, purification, and identification of the liver canalicular ecto-ATPase as NTPDase8. Am. J. Physiology-Gastrointestinal Liver Physiol. 292 (3), G785–G95 (2007).
Lecka, J., Rana, M. S. & Sévigny, J. Inhibition of vascular ectonucleotidase activities by the pro-drugs ticlopidine and clopidogrel favours platelet aggregation. Br. J. Pharmacol. 161 (5), 1150–1160 (2010).
Kukulski, F. et al. Comparative hydrolysis of P2 receptor agonists by NTPDases 1, 2, 3 and 8. Purinergic Signal. 1, 193–204 (2005).
Adzic, M. & Nedeljkovic, N. Unveiling the role of ecto-5′-nucleotidase/CD73 in astrocyte migration by using pharmacological tools. Front. Pharmacol. 9, 153 (2018).
Zhang, Y. et al. Discovery and optimization of betulinic acid derivatives as novel potent CD73 inhibitors. Bioorg. Med. Chem. 59, 116672. https://doi.org/10.1016/j.bmc.2022.116672 (2022).
Scaletti, E., Huschmann, F. U., Mueller, U., Weiss, M. S. & Sträter, N. Substrate binding modes of purine and pyrimidine nucleotides to human ecto-5′-nucleotidase (CD73) and inhibition by their bisphosphonic acid derivatives. Purinergic Signal. 17 (4), 693–704. https://doi.org/10.1007/s11302-021-09802-w (2021).
Release, S. 4: Desmond molecular dynamics system (DE Shaw Research, 2017).
Acar, Ö. Ö. et al. A Small Indole Derivative Isolated From Caper (Capparis ovata) as an Inducer of P53-Mediated Apoptosis in Prostate Cancer: Comprehensive In Vitro and In Silico Studies. J. Biochem. Mol. Toxic. 40 (1), e70666. https://doi.org/10.1002/jbt.70666 (2026).
Aftab, H. et al. A series of 4-thiomorpholinophenyl-thiosemicarbazones as cholinesterase inhibitors with anti-neuroblastoma effects. Bioorg. Chem. 168, 109364. https://doi.org/10.1016/j.bioorg.2025.109364 (2026).
Tokalı, F. S., Şenol, H., Ateoglu, Ş. & Akbaş, F. A series of quinazolin-4(3H)-one-morpholine hybrids as anti-lung-cancer agents: Synthesis, molecular docking, molecular dynamics, ADME prediction and biological activity studies. Chem. Biol. Drug Des. 104 (1), e14599. https://doi.org/10.1111/cbdd.14599 (2024).
Tokalı, F. S., Şenol, H., Ateşoğlu, Ş., Tokalı, P. & Akbaş, F. Exploring highly selective polymethoxy fenamate isosteres as novel anti-prostate cancer agents: Synthesis, biological activity, molecular docking, molecular dynamics, and ADME studies. J. Mol. Struct. 1319, 139519. https://doi.org/10.1016/j.molstruc.2024.139519 (2025).
Tokalı, F. S. et al. Novel phenolic Mannich base derivatives: synthesis, bioactivity, molecular docking, and ADME-Tox Studies. J. Iran. Chem. Soc. 19 (2), 563–577. https://doi.org/10.1007/s13738-021-02331-8 (2022).
Kılınç, N., Açar, M., Tuncay, S. & Karasakal, Ö. F. Identification of Potential Inhibitors for Severe Acute Respiratory Syndrome-related Coronavirus 2 (SARS-CoV-2) Angiotensin-converting Enzyme 2 and the Main Protease from Anatolian Traditional Plants. Lett. Drug Des. Discov. 19 (11), 996–1006. https://doi.org/10.2174/1570180819666211230123145 (2022).
Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University, Saudi Arabia, through Large Research Project under grant number for the year 1447. J.I is thankful to the Higher Education Commission of Pakistan (HEC) via NRPU project no 20–15846/NRPU/R&D/HEC/2021–2020 and China Pakistan Economic Corridor-Collaborative Research Grant # P2–345. J.S. received support from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2023-05498). Institutional support for J.S. was provided by the Fonds de recherche du Québec.
Funding
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University, Saudi Arabia, through Large Research Project under grant number for the year 1447. J.S. received support from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2023-05498). Institutional support for J.S. was provided by the Fonds de recherche du Québec.
Author information
Authors and Affiliations
Contributions
Zahra Batool, Hina Aftab, Mostafa A. Ismail: synthesis and spectral analysis. Mariya al-Rashida, Talha Islam, and Ajmal Khan: molecular docking, MD simulation. Shireen Mona Dutt, Jamshed Iqbal: biological assays. Nicolly Espindola Gelsleichter: preparation of recombinant enzymes. Julie Pelletier: preparation of recombinant enzymes and edition, Jean Sévigny: providing recombinant enzymes, review & editing. Rima D. Alharthy: Formal analysis, data interpretation. Halil Şenol: Formal analysis, methodology, data interpretation, software, writing-review original draft and review & editing. Zahid Shafiq, Jamshed Iqbal: conceptualization, supervision, writing-review original draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Batool, Z., Dutt, S.M., al-Rashida, M. et al. Synthesis, biological evaluation and molecular docking studies of N-propylsulfonyl indole-linked hydrazinecarbothioamides as selective ecto-5′-nucleotidase and NTPDase inhibitors. Sci Rep 16, 14419 (2026). https://doi.org/10.1038/s41598-026-50728-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-50728-3
