
Abstract
Collaborative review of molecular profiling data by multidisciplinary molecular tumor boards (MTB) is increasingly important for improving patient management and outcomes, though currently relies nearly exclusively on nucleic acid next-generation sequencing (NGS) and limited panels of immunohistochemistry-based protein abundance data. We examined the feasibility of incorporating real-time laser microdissection (LMD) enrichment of tumor epithelium and commercial CLIA-based reverse phase protein array (RPPA) protein drug target expression/activation profiling into our cancer center’s MTB to complement standard clinical NGS-based profiling. The LMD-RPPA workflow was performed within a therapeutically permissive timeframe with a median dwell time of nine days, during which specimens were processed outside of standard clinical workflows. The RPPA-generated data supported additional and/or alternative therapeutic considerations for 54% of profiled patients following review by the MTB. These findings suggest that integrating proteomic/phosphoproteomic and NGS-based genomic data creates opportunities to further personalize clinical decision-making for precision oncology.
Similar content being viewed by others


Introduction
Cancer diagnostics and treatments have significantly improved in recent years due in part to the continuous development and optimization of highly sensitive technologies and molecular assays for improved early detection, routine screening, and therapeutic selection; collectively contributing to advancing personalized medicine approaches. Despite these advances, cancer remains the second leading cause of death in the United States, with over two million new cancer diagnoses and 611,720 cancer-related deaths predicted to occur in 20241. Molecular Tumor Boards (MTBs) are becoming more commonplace in clinical oncology, in which multidisciplinary committees comprised of clinical and research specialists review the clinical histories and molecular profiling data for each patient and identify the best therapeutic options available, evidenced by clinical trials or known tumor molecular signatures.
The molecular profiling data available from clinical trials is largely from next-generation sequencing (NGS) studies correlating genomic and/or transcriptomic alterations with therapeutic responsiveness. Cancer therapeutics, however, largely exert their anti-cancer activity at the level of the proteome, either by targeting specific protein activities (e.g., trastuzumab targeting human epidermal growth factor receptor 2 (HER2) kinase signaling2), or by disrupting cellular processes mediated by proteins (e.g., gemcitabine inhibiting DNA synthesis and cell cycle progression3). Genomic variation and transcriptomic expression are at best loosely correlated with protein activity and/or abundance4,5,6,7, attributed partially to post-translational modifications (PTMs) that modify protein activity, the presence of multiple protein isoforms transcribed/translated from a single gene, and the post-translational stability and/or localization of a protein either within or outside of a cell. Currently, protein-based profiling in clinical oncology is essentially limited to diagnostic immunohistochemistry (IHC), despite the fact that quantitation of protein activation (i.e. phosphorylation) is often predictive of therapeutic response8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31.
The translational (clinical) relevance of transcript-based signatures is often limited in part due to the fact that large-cohort studies predominantly identify signatures from bulk tissue samples containing variable admixtures of tumor, stroma, and immune cells present within the tumor microenvironment (TME)32,33, which complicates therapeutic selection. Recent provocative studies have demonstrated widespread biological “misinterpretation” of gene expression signatures due to cell admixture34, and include the identification of genes/proteins correlating with poor outcomes in cancer patients that are stromal derived5,35,36,37,38,39. Tissue preparatory techniques that isolate tumor and/or non-tumor cell populations, such as laser microdissection (LMD) or single cell-based analyses (single-cell sequencing or single cell proteomics), are needed to selectively and accurately identify cell-specific prognostic and/or theragnostic molecular profiles from the tumor tissue microenvironment (TME).
We recently demonstrated that incorporating CLIA-based reverse phase protein array (RPPA) drug target mapping into a precision oncology MTB for therapeutic decision-making significantly increases both actionability frequency and patient outcomes9,10. Based on the promise of RPPA and/or other new multiplexed and quantitative proteomic platforms, which directly measure the expression and activation of actionable protein drug targets, we sought to build on this work by evaluating the operational feasibility of integrating our hyphenated LMD-RPPA CLIA workflow for a real-time multi-omic return of data in our cancer center’s precision oncology MTB.
Results
Clinical characteristics of cohort
A total of 174 patients with solid tumor malignancies seeking treatment were consented and enrolled in this study to examine the feasibility of incorporating a hyphenated workflow involving LMD enrichment of tumor epithelium for RPPA-based proteomic analysis (Fig. 1, Supplementary Table 1). All patients were from an intention-to-treat population; therefore, the primary aim of this study was to determine whether the implementation of new multi-omic workflows into MTB discussions could be performed within a treatment-permissible timeframe and to secondarily determine whether complementary RPPA-based data would reveal additional or alternative therapeutic options based on NGS and IHC data alone.

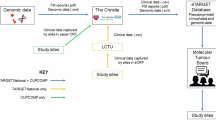
A Flowchart depicting the number of patients included or excluded at each timepoint of the study workflow. B Representative timeline and pre/post-LMD images. Purple arrows represent the days in which the specimen block was in the possession of the proteomic workflow team, i.e., outside the normal clinical MTB workflow.
Our cohort comprises 31 different primary malignancies, broadly categorized into 9 disease groups and 41 different histological cell types (Table 1). Most patients had gastrointestinal (n = 77 patients, 44.3%), breast (n = 26 patients, 14.9%), or skin (n = 23 patients, 13.2%) tumors. Most tumors were adenocarcinomas (n = 87 patients, 50%), though several tumors of rare histology were included, such as angiosarcomas (n = 2), ganglioneuroma (n = 1), inflammatory myofibroblastic tumor (IMT, n = 1), leiomyosarcoma (n = 1), Merkel cell carcinomas (n = 2), melanocytic matrical carcinoma (n = 1), pigmented dermatofibrosarcoma protuberans (“Bednar tumor”, n = 1), and sebaceous carcinoma (n = 1).
Patient dropouts and censors
Patients were excluded during the feasibility assessment if all available surgical specimens had insufficient tumor tissue to support LMD enrichment (n = 54 patients) prior to RPPA analysis, if the patient died during the feasibility assessment period (n = 4 patients), and/or prior to MTB review of the multi-omic data (n = 4 patients), or if the relevant biopsy/surgery was performed at an external facility from which the tissue specimen or slides sectioned for LMD were unable to be obtained for analysis (“unavailable external specimen transfer”, n = 4 patients) (Fig. 1B, Table 2). Specifically, 59 (34%) of these patient failures were related to the inability to complete RPPA due to the aforementioned insufficient quantity of tissue or quality of LMD protein sample. Two patients (who were not lung cancer patients) were excluded when NGS was completed prior to RPPA analysis (“protocol non-adherence”). Further, despite achieving the target LMD harvest area (5–10 mm2 enriched tumor), five patients were excluded because the protein lysate concentration was insufficient for RPPA analysis. Three patients had multiple concurrent primary sites of disease, from each of which specimens were considered for analysis.
We sought to evaluate if there were associations between select tumor types and rates of patient dropout in our multi-omic workflow. Given the heterogeneous tumor origins observed in this pan-cancer study, only primary sites of disease with more than four patients were included in this assessment (Table 2). Among these, patients with primary tumors originating in the lung (9/12 patients, 75%), stomach (6/10 patients, 60%), and pancreas (18/33 patients, 55%) had the highest dropout rates. The lowest dropout rates were in patients with esophageal (0/4 patients, 0%), rectal (1/7 patients, 14%), or breast (8/26 patients, 31%) cancer. The most frequent reason for patient dropout was due to inadequate tissue availability for multi-omic analysis. No suitable tumor tissue specimens were available for NGS from two patients, so liquid biopsies (Tempus, Inc.) from these patients were profiled by NGS.
Alternative specimens for RPPA were selected when the amount of tissue available within a specimen block was insufficient (i.e. the necessary amount of tissue sections to achieve the target tumor LMD area could not be generated without exhausting the specimen). Among the inadequate tissue specimens, those obtained from lung (n = 26 cassettes), lymph nodes (n = 21 cassettes), liver (n = 18 cassettes), pancreas (n = 13 cassettes), and prostate (n = 12 cassettes), were most commonly collected via fine needle aspirates (FNA) and had the highest rates of failure due to either minimal tissue depth within the specimen block or minimal tumor area for LMD enrichment (Supplementary Table 2). Of note, not all biopsies from these organs failed, as LMD-enriched samples from lung, liver, pancreas, and prostate specimens were successfully obtained from other patients and analyzed in this study.
Multi-omic characterization of tumors
Tumor epithelium was selectively harvested by LMD from 110 patients for quantitative proteomic analysis by RPPA, from which 105 successful RPPA and NGS reports were generated. The same tissue specimens were prioritized for multi-omic analysis by RPPA and NGS (using bulk tissue samples) whenever possible (Table 3). Matched specimens (i.e., same FFPE block) were used for 72/105 patients for RPPA and NGS analysis. Tissues from unmatched (different) specimen blocks were used for analysis by NGS vs. RPPA from 33 patients. RPPA was used to quantify the abundance of 32 proteins and/or phosphoproteins with known cancer significance and/or that represent actionable drug targets40,41 (Supplementary Table 3). The RPPA and NGS reports from 101 patients were reviewed by the MTB. The multi-omic results from patients who died while the RPPA and/or NGS analyses were ongoing (4/105, 3.8%) were not reviewed by the MTB.
Following specimen receipt by the proteomics team (median day 14 after patient consent; Supplementary Table 1), a representative H&E tissue section was reviewed and the tumor areas for LMD harvest were annotated and confirmed by a board-certified pathologist (median day 16 after patient consent; day 6 of the proteomic workflow). Specimens that passed this feasibility assessment (i.e. had sufficient areas of tumor to support LMD enrichment for RPPA and NGS) were further sectioned onto polyethylene naphthalate (PEN) membrane slides. The proteomic arm of the study required possession of each tissue specimen for a median of 9 days (day 24 after patient consent), during which time the specimen was not available to the hospital or NGS companies for standard clinical testing. Specimen blocks were returned to the hospital for NGS after generating tissue sections for LMD-RPPA or after failing the feasibility assessment. After tissue sectioning, the LMD-RPPA and NGS analyses occurred essentially in parallel (representative days 24–41 after patient consent), with the exception of patients with lung cancer or for specimens that failed the feasibility assessment for proteomic analysis and were therefore exclusively reserved for NGS analysis (Supplementary Table 1, Supplementary Table 4). NGS reports were available within a median of 11 days (representative day 35 after patient consent) after receipt of the FFPE tissue specimen by Tempus (Supplementary Table 4). RPPA reports were available within a median of 8 days (representative day 41 after patient consent) after receipt of the LMD sample lysate by Ignite Proteomics (Supplementary Table 1).
Given the heterogeneity of patients in this pan-cancer patient cohort, a preliminary analysis examining tumors associated with the highest and lowest protein recovery (µg/mm2 LMD tumor area) was performed. The median recovery across all samples was 0.574 µg/mm2 (Supplementary Table 5). Across tumor types, the highest protein recoveries were from tumors originating in the sebaceous gland (median = 1.86 µg/mm2, n = 2), neuroendocrine tumors (1.51 µg/mm2, n = 1), and the gastroesophageal junction (1.21 µg/mm2, n = 1), while the lowest recoveries were from tumors from the uterus (0.08 µg/mm2, n = 1), lung (median = 0.28 µg/mm2, n = 4), and stomach (median = 0.28 µg/mm2, n = 6) (Supplementary Fig. 1A). When categorizing the data by tissue sites sampled, biopsies obtained from the eyelid and conjunctiva (2.14 µg/mm2, n = 1), omentum (median = 1.33 µg/mm2, n = 2), and colon (median = 1.29 µg/mm2, n = 2) had the highest recoveries, while those from the endometrium (0.08 µg/mm2, n = 1), stomach (median=0.19 µg/mm2, n = 3), and epiploic appendage (0.22 µg/mm2, n = 1) had the lowest recoveries (Supplementary Fig. 1B). Finally, when categorizing the data by tumor histology, neuroendocrine tumors (1.51 µg/mm2, n = 1), gastrointestinal stromal tumors (GIST; median = 1.34 µg/mm2, n = 2), and pigmented epithelioid melanocytoma (1.28 µg/mm2, n = 1) had the highest protein recovery, whereas leiomyosarcoma (0.08 µg/mm2, n = 1), signet ring cell carcinoma (0.22 µg/mm2, n = 1), and ductal adenocarcinoma (0.23 µg/mm2, n = 1) had the lowest protein recoveries (Supplementary Fig. 1C).
Proteome-informed clinical considerations
The multi-omic data from NGS and RPPA were reviewed by our cancer center’s MTB for 101 patients to generate a personalized list of considerations for later-line therapy based on currently available targeted therapies and clinical trials. The expression and/or activation levels for each of the 32 RPPA analytes were reported as a continuous percentage ranging from non-expressed/non-activated (0%) to highly expressed/highly activated (100%) and as a normalized intensity integer representing normalized percentage quartiles as follows: 0 (non-expressed/non-activated), 1+ (low expression/activation), 2+ (moderately expressed/activated), or 3+ (highly expressed/activated) (Fig. 2, Supplementary Table 6), as previously described11,41. Recommendations were based on known mechanisms of action from the FDA-approved package insert for each drug, combined with levels of evidence of each biomarker derived from publications where the marker was found to be predictive of response using human clinical tissue samples obtained prior to treatment in human clinical trials where outcome was known. Broadly, highly abundant (2+ or 3+) RPPA targets and/or select less abundant (1+) RPPA targets predictive of a select therapeutic based on clinical trial outcomes were prioritized for a patient’s treatment regimen.

Heatmap depicting the RPPA-quantified expression and/or activation of 32 protein and phosphoprotein analytes with known relevance as druggable targets in solid tumors relating to androgen signaling, cell cycle regulation, cellular proliferation, DNA damage response, EGFR/HER2 signaling, immune checkpoints, oncogenic RTK signaling, PI3K/AKT/mTOR signaling, RAS/MEK/ERK signaling, stem cell signaling, and the tumor microenvironment. NA/QNS = not applicable or quantity not sufficient, for RPPA abundances that were not reported for a given patient.
Across all disease sites, the RPPA-derived data supported additional or alternative therapeutic modalities for 55/101 patients (54.5%) (Fig. 2, Supplementary Table 6). Prioritizing groups with ≥4 successfully reviewed patients, the primary sites of disease with the highest rates of recommended changes to therapy included breast (13/18 patients, 72.2%), skin (7/11 patients, 63.6%), and pancreas (9/15 patients, 60%). Patients with prostate (1/5 patients, 20%), soft tissue (1/4 patients, 25%), and rectal (2/6 patients, 33.3%) tumors were least likely to have alternative therapies recommended based on the RPPA workflow and/or the specific antibody panel used for analysis.
At the time of publication, the recommended changes had not yet been implemented because most patients had not exhausted their standard-of-care therapeutic options. Patient outcomes that result with/without implementation of the RPPA-informed changes will be evaluated and presented in the future.
Recommendations for integrating proteomic workflows into clinical practice
The most common limitations encountered while piloting this integrated multi-omic clinical MTB workflow are described in Table 4. Miniscule tissue volumes within specimen blocks and/or low tumor cellularity of the biopsied tissue (i.e., “inadequate tissue specimen(s)”) most frequently prevented proteomic/multi-omic analysis. The amount of tissue needed for each workflow (RPPA, DNA-seq, and/or RNA-seq) and a contextual representation of what that physically equates to in clinical practice per disease and biopsy tissue source should be carefully communicated between the research and surgical/interventional radiology teams prior to the adoption of proteomic workflows into MTBs and/or prior to surgery/biopsy. If the pre-biopsy tumor size permits and if clinically safe for the patient, additional tissue collections to obtain multiple biopsy cores, as well as either embedding all cores into a single FFPE specimen block or pooling of tissue from individual cores embedded into multiple blocks, may help reduce the number of specimens that dropout due to inadequate tissue availability.
Care should be taken during histopathology sectioning when generating tissue sections, particularly for specimens with minimal starting material, such as thin needle biopsies. Specifically, while “facing” the tissue specimen (i.e., shaving off the top layers of the FFPE block to obtain a full tissue section representing the complete intact cross-sectional area), histopathology technicians should set the microtome blade to a minimum thickness to minimize the loss of tissue.
Discussion
Genomic variation and transcriptomic expression are imperfect and indirect proxies of protein drug target expression and/or activation. Accurate and sensitive quantitative and multiplexed measurements of proteins and phosphoproteins are increasingly being acknowledged as a missing component of molecularly informed treatment planning. Moreover, recent studies have shown that pre-treatment levels of tumor proteins and phosphoproteins are highly predictive of response in the absence of and independent of underpinning genomic alterations11,12,13,14,15,16,17,18,42,43,44. A critical need thus exists to identify and validate proteomic and/or phosphoproteomic biomarkers with diagnostic, prognostic, predictive, and/or theragnostic importance, which may only occur through the incorporation of proteome-focused analyses into clinical practice. We, therefore, present our experience incorporating real-time proteomic testing via RPPA analysis into our cancer center’s MTB workflow, for which we demonstrate alignment of proteomic/multi-omic analyses within a treatment-permissible timeframe and the potential for significant clinical benefit. Specifically, collaborative review of the proteomic data in MTB discussions resulted in the identification of additional and/or alternative treatment considerations for 54% of the patients who had been successfully profiled and only prolonged standard clinical workflows by a median of nine days.
Integrating our hyphenated LMD-RPPA workflow into our cancer center’s precision medicine approach facilitated several anecdotal discoveries with profound clinical significance. One patient was diagnosed by IHC and standard clinical diagnostics with an anaplastic lymphoma kinase (ALK)-positive inflammatory myofibroblastic tumor41. RPPA-derived proteomic/phosphoproteomic data on the LMD-enriched tumor revealed persistent EGFR signaling in both the primary and recurrent tumors, which was exclusively detected by RPPA, with no corresponding genomic alterations detected by NGS. ALK signaling was not activated (i.e., not phosphorylated) in the patient’s tumor after treatment with an ALK-directed tyrosine kinase inhibitor (TKI), thereby contraindicating the continued use of subsequent generations of ALK TKI. This indication was further supported by the presence of a secondary ALK mutation that was correlated with reduced ALK TKI sensitivity in vitro.
Another patient in this study had recurrent triple-negative breast cancer (TNBC) and progressed on five prior lines of therapy prior to RPPA analysis8. Contrary to the IHC-derived evidence as being HER2-negative (IHC score 0), the RPPA data from the LMD-enriched tumor specimen revealed substantial HER2Total and phosphorylated (p)HER2Y1248 and pHER3Y1289 levels, which supported the use of a HER2-directed therapy. The patient had an excellent pathologic complete response (pCR) to 6th-line treatment with trastuzumab deruxtecan (T-DXd), a therapy that would have not been otherwise selected based on standard clinical diagnostics alone. Specifically, after four cycles of T-DXd the patient had near-resolution of her hepatic lesions and, after nine cycles, had no measurable disease, including no new brain metastases.
The LMD-enriched tumor samples from several patients with pancreatic adenocarcinoma (PDAC) enrolled in this study were found to have elevated HER2Total expression, comparable to the levels observed in breast cancer patients with HER2-positive and HER2-low tumors45. While not significantly activated (phosphorylated), the high rate of elevated HER2Total expression in PDAC tumors provided foundational evidence supporting further studies examining the use of HER2-targeting antibody–drug conjugates (ADCs) in this patient population, for whom existing therapeutic options are limited and clinical outcomes are dismal. Further, observations of protein expression and/or activation patterns that diverge from current or dogmatic understanding, such as this finding, may support a need for selective RPPA panels that align with known disease biology per cancer type into MTB proteomic/multi-omic workflows.
While the survival outcomes for cancer patients continue to gradually improve overall, current estimates predict only a 69% 5-year survival rate for all cancers, and the outcomes for patients with pancreatic, liver, esophageal, and lung cancers remain far poorer46. There is no specific efficacy threshold required for FDA approval of new drugs or new drug indications. The median response rate endpoint for approval for recent drugs and/or indications is ~41%, with a median response duration of only 9 months47. These low efficacy rates critically underscore the necessity for better therapeutic options.
Our data support the findings of recent studies such as the I-SPY 2 trial42,43 and the SideOut trials9,10 to consider treating malignancies as “molecular target-oma’s” based on multi-omic profiling data (to specifically include proteome/phosphoproteome-level characterization of enriched tumor cell populations) in a personalized medicine manner, rather than strictly on the basis of disease origin site and histology. Specifically, the interpatient heterogeneity in our study observed supports the need for treating each patient as an “n-of-1” using individualized molecular profiling for precision therapeutic decision-making.
The SideOut 2 trial evaluated CLIA-based RPPA analysis as a component of multi-omic profiling for patients with refractory metastatic breast cancer and found that 56% of patients who received treatment informed by their personal molecular profiling data demonstrated positive clinical improvement10. The I-SPY 2 trial was similarly examined using personalized RPPA profiling data and the preferential assignment of patients to treatment arms through an adaptive randomization strategy informed by patient-specific molecular profiling in combination with continuously updated response probabilities calculated from patients with similar molecular signatures42. Many other research investigations and clinical trials are increasingly recognizing the need for and potential benefit of quantitative protein and phosphoprotein biomarkers for improving clinical outcomes8,9,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31.
We successfully demonstrate that incorporating real-time LMD enrichment of tissue specimens and subsequent RPPA analysis as an orthogonal dataset to complement NGS data from bulk tissue specimens can be performed within a clinically permissive timeframe and often yields additional and/or alternative therapeutic options when the multi-omic data are collectively reviewed in the MTB setting.
Methods
Patient cohort
From October 2021 to December 2023, a total of 174 adult (≥18 years of age) patients with advanced solid tumor malignancies seeking treatment were consented and enrolled under a Western IRB-approved protocol into the Inova Schar Cancer Institute’s Molecular Tumor Board (MTB) study (protocol number U20-19-4308). The written informed consent included the provision to analyze and publish information and data regarding the patients’ clinical presentation and results and data from precision medicine/next-generation genomic sequencing testing, including the RPPA profiling data. Eligible patients had pathology-confirmed cancer by tumor biopsy and had tumor available for testing. Patients with multiple concurrent malignancies were eligible. Patients had received varied lines of previous therapy, ranging from treatment-naive to heavily pre-treated. All experimental protocols involving human data in this study were in accordance with the Declaration of Helsinki and written informed consent was obtained from all patients.
A total of 262 formalin-fixed, paraffin-embedded (FFPE) surgically resected and/or biopsied tissue specimens were obtained from patients (average 2 blocks/patient, range 0–10). The specimens were acquired through surgical resections, excisions, computed tomography (CT)-guided biopsies, fine needle aspirations (FNA), punch biopsies, and/or bone biopsies. Liquid biopsies were used exclusively for NGS if all available solid tissue specimens were of insufficient quality and/or quantity.
Associated costs per specimen for in-house histology and LMD tissue processing, as well as RPPA analysis, were covered by the study at no cost to the patient. Costs associated with NGS were billed to the patient as a part of their standard of care treatment, with costs varying based on the specific assays ordered, insurance coverage, and financial assistance.
Feasibility assessment
After initial clinical diagnostic testing, the tissue specimens were assessed to determine the total amount of available tissue within a specimen block (width and depth of tissue) and the amount of specific tumor area available for enrichment via LMD. This determination was used to prioritize specimens for real-time quantitative analysis via a commercially available CLIA RPPA-based assay that measured 32 protein/phosphoprotein drug targets40,41,45 followed by real-time NGS for DNA-seq and/or RNA-seq.
To assess feasibility, a representative hematoxylin and eosin (H&E)-stained thin section for each case was scanned using the Aperio ScanScope XT slide scanner (Leica Microsystems). Foci of target tumor epithelium on the representative H&E slide were precisely annotated in Aperio eSlide Manager and Aperio ImageScope (Leica Microsystems) and confirmed by a board-certified pathologist. Specimens that contained sufficient tumor area and tissue depth within the FFPE block were credentialed for downstream workflows. Subsequent serial sectioning was performed with the highest priority of allocating tissue for NGS and then RPPA. Chronologically (based on pre-defined optimal workflow outlined in the study protocol), tissue sections were first generated and allocated for the quantitative proteomic and phosphoproteomic workflows (RPPA), after which the specimen blocks were shipped to Tempus Labs, Inc. (Chicago, IL, USA) for NGS. Per institution preference, specimens from patients with lung cancer went directly from biopsy to NGS (prior to proteomic feasibility assessment) to accelerate treatment-based decision-making for administering EGFR inhibitors as indicated. Specimens that failed the feasibility assessment for LMD enrichment prior to RPPA-based proteomic analysis (i.e., had insufficient target tumor area per section and/or tissue thickness) but had sufficient tissue for NGS alone were shipped to Tempus and an alternative specimen block was requested, if available, for additional review for inclusion in the RPPA workflow. Specimens that failed the feasibility assessment for both the proteomic and/or NGS workflows were returned immediately, without testing being performed.
Tissue specimen preparation for laser microdissection
A total of 111 FFPE tissue specimens were sectioned (8 µm) onto PEN membrane slides for LMD enrichment. The number of total slides was calculated from the number of sections needed to obtain a total cross-sectional area of enriched tumor epithelium of 5–10 mm2 for RPPA (median 2 slides/block, range 1–11, n = 111 specimens). All slides were stained prior to LMD enrichment as previously described5. Briefly, slide preparation for LMD prior to RPPA did not involve counter-staining with eosin after hematoxylin but included color development in Scott’s Tap Water (Thermo Fisher Scientific, Inc.) and two final drying steps in xylenes. One representative LMD tissue section per specimen was imaged before and after LMD for each workflow for quality assurance and quality control (QA/QC) purposes.
Laser microdissection
LMD enrichment of tumor epithelium was performed on the LMD7 (Leica Microsystems). LMD tumor samples for RPPA analysis were collected in real-time (as patients consented to the study and the specimens passed the feasibility assessment) into cylindrical pressure cycling technology (PCT, Pressure Biosciences, Inc.) microtubes. A lysis/extraction buffer was added at a ratio of 2.5 µl buffer per mm2 LMD tissue, as previously described45. The PCT microtubes were capped with PCT microcaps and placed into 0.5 ml PCR tubes, briefly centrifuged, and stored at −80 °C until sample lysis.
Reverse phase protein microarray
LMD-enriched tumor samples destined for RPPA analysis were heated at 95 °C for 20 min, briefly centrifuged, and heated at 80 °C for 2 h. After heating, the tubes were stored at 4 °C for 1 min and then centrifuged for 14,000 rpm for 15 min. The lysate supernatants were transferred to fresh low protein binding tubes and stored at −80 °C until shipment on dry ice to Ignite Proteomics (Golden, CO, USA) for RPPA analysis.
A 32-marker, CLIA-based RPPA analysis was performed as previously described8,48 to examine expression and activation (phosphorylation) of well-known actionable protein drug targets with known clinical relevance in solid tumors40,41,45. Validation criteria for the CLIA-accredited RPPA assay are provided in Supplementary Note 1. Analytes on the RPPA panel were selected based upon the known mechanism of action of these FDA-targeted therapeutics as directly reported from section 12 (“Clinical Pharmacology, Mechanism of Action”) of the FDA-approved package insert. Sample lysates were heated and robotically arrayed onto nitrocellulose-backed slides that were probed with primary antibodies and a biotinyl tyramide amplification system to generate a fluorescent signal proportional to the analyte abundance. Signals were normalized to total protein and compared to a reference population to generate percentile and quartile scores in a patient report format. A minimum sample concentration of 100 µg/ml (total: 5 µg protein in 50 µl volume) was required for quantification by RPPA. Per analyte, 10 nl of sample lysate (~1 ng) was used for printing the microarrays. Multiple depositions were made to print samples with concentrations below this threshold, when possible.
Next generation sequencing
Commercially available NGS was performed by Tempus Labs, Inc. DNA-seq was performed using the Tempus xT 648 gene panel49. Additional NGS testing (i.e., RNA-seq) was performed per physician discretion (Tempus xF 105 gene panel for cell-free DNA (cfDNA)50, PD-L1, tumor origin, etc.) or diagnosis. The samples for DNA-seq were prepared from tissue sections with an estimated 52% median tumor cellularity (range = 20–90%) after macrodissection/microdissection.
Molecular Tumor Board Review
The NGS profiling data for each patient, along with demographic metadata including disease, age, and sex, was initially reviewed to evaluate therapeutic treatment considerations at our cancer center’s weekly Molecular Tumor Board (MTB) meeting attended by medical oncologists, molecular pathologists, and clinical trials research staff. After the initial NGS review, the RPPA-based proteomics and NGS results were reviewed together with the relevant clinical histories to arrive at therapeutic considerations for each patient by the MTB. The MTB discussions were held biweekly and were attended in a hybrid virtual and in-person format by specialists from the clinical and research teams. The clinical team consisted of board-certified medical oncologists and pathologists, medical oncology fellows, genetic counselors, and clinical trials research staff. The research team consisted of subject-matter expert scientists specializing in molecular profiling (proteomics, phosphoproteomics, genomics, and/or transcriptomics), as well as histopathology and research technicians. A minimum of two board-certified medical oncologists were required to discuss the clinical results, interpret the multi-omic profiling data, and provide an MTB-informed consensus treatment recommendation based on the molecular profiling results. The clinical history and molecular profiling reports for each patient were first presented individually to the MTB in a lecture-style format led by a board-certified medical oncologist, after which the discussion was opened to a roundtable format to facilitate input and open dialog between members of the clinical and research teams. After discussion and interpretation of the multi-omic data, an MTB-informed consensus treatment recommendation was recorded, provided to the treating physician, and included in patients’ electronic medical records, and it was documented if there was a change from the original MTB NGS-informed treatment recommendations.
Bioinformatic and statistical analysis
Descriptive statistics were used to summarize the clinical characteristics of the patients. Turnaround time metrics were calculated using 179 specimens obtained from 119 unique patients unless otherwise specified. Specimens were excluded from all or some of the turnaround time calculations for one or more of the following reasons, as appropriate: (1) the patient was pre-defined as a “lead-in case” to facilitate early workflow transfers, (2) the specimen acquisition and/or analytical workflows for the patient did not adhere to the intended chronological workflow schema (e.g., NGS was performed prior to RPPA), (3) additional specimens were requested for clinical and/or research-related reasons (e.g., to examine pre- vs. post-treatment changes), (4) the related workflow step was hindered by reliance on a single vacant staffing position, or (5) the RPPA analysis was impacted by a universal commercial shortage of one of the required reagents and could not be performed until the supply issue was resolved. Box-and-whisker plots were generated using BoxPlotR51. The RPPA abundance/activation level heatmap was generated in R (version 4.4.0) with ggplot2 (version 3.5.1) and patchwork (version 1.2.0).
Data availability
The NGS data that support the findings of this study were generated by Tempus Labs, Inc. However, restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are available from the authors upon reasonable request and with the permission of Tempus Labs, Inc. The RPPA abundance data (normalized intensity integers and continuous percentiles) is available in Supplementary Table 6. The RPPA antibody signal intensity data used to generate the proteomic/phosphoproteomic abundance data is available upon request.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA: Cancer J. Clin. 74, 12–49 (2024).
Goldenberg, M. M. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin. Ther. 21, 309–318 (1999).
Hertel, L. W. et al. Evaluation of the antitumor activity of gemcitabine (2’,2’-difluoro-2’-deoxycytidine). Cancer Res. 50, 4417–4422 (1990).
Gonçalves, E. et al. Widespread post-transcriptional attenuation of genomic copy-number variation in cancer. Cell Syst. 5, 386–398.e384 (2017).
Hunt, A. L. et al. Extensive three-dimensional intratumor proteomic heterogeneity revealed by multiregion sampling in high-grade serous ovarian tumor specimens. iScience 24, 102757 (2021).
Burdett, N. L. et al. Multiomic analysis of homologous recombination-deficient end-stage high-grade serous ovarian cancer. Nat. Genet. https://doi.org/10.1038/s41588-023-01320-2 (2023).
Edfors, F. et al. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 12, 883 (2016).
Johnston, L. E. et al. Proteomics based selection achieves complete response to HER2 therapy in HER2 IHC 0 breast cancer. npj Precis. Oncol. 8, 203 (2024).
Jameson, G. S. et al. A pilot study utilizing multi-omic molecular profiling to find potential targets and select individualized treatments for patients with previously treated metastatic breast cancer. Breast Cancer Res. Treat. 147, 579–588 (2014).
Pierobon, M. et al. Multi-omic molecular profiling guide’s efficacious treatment selection in refractory metastatic breast cancer: a prospective phase II clinical trial. Mol. Oncol. 16, 104–115 (2022).
Wulfkuhle, J. D. et al. Evaluation of the HER/PI3K/AKT family signaling network as a predictive biomarker of pathologic complete response for patients with breast cancer treated with neratinib in the I-SPY 2 TRIAL. JCO Precis. Oncol. 1–20 https://doi.org/10.1200/po.18.00024 (2018).
Albain, K. S. et al. Neoadjuvant trebananib plus paclitaxel-based chemotherapy for stage II/III breast cancer in the adaptively randomized I-SPY2 trial—efficacy and biomarker discovery. Clin. Cancer Res. 30, 729–740 (2024).
Wolf, D. M. et al. Mechanism of action biomarkers predicting response to AKT inhibition in the I-SPY 2 breast cancer trial. npj Breast Cancer 6, 48 (2020).
Shi, Z. et al. Functional mapping of AKT signaling and biomarkers of response from the FAIRLANE trial of neoadjuvant ipatasertib plus paclitaxel for triple-negative breast cancer. Clin. Cancer Res. 28, 993–1003 (2022).
Abu-Khalaf, M. M. et al. AKT/mTOR signaling modulates resistance to endocrine therapy and CDK4/6 inhibition in metastatic breast cancers. npj Precis. Oncol. 7, 18 (2023).
Wang, F. et al. Phosphorylated EGFR expression may predict outcome of EGFR-TKIs therapy for the advanced NSCLC patients with wild-type EGFR. J. Exp. Clin. Cancer Res. 31, 65 (2012).
Treilleux, I. et al. Translational studies within the TAMRAD randomized GINECO trial: evidence for mTORC1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer. Ann. Oncol. 26, 120–125 (2015).
Gonzalez-Ericsson, P. I. et al. Tumor-specific major histocompatibility-ii expression predicts benefit to anti-PD-1/L1 therapy in patients with HER2-negative primary breast cancer. Clin. Cancer Res. 27, 5299–5306 (2021).
Parsons, B. M., Meier, D. R., Gurda, G. T., Lofgren, K. A. & Kenny, P. A. Exceptional response to crizotinib in an MET-amplified triple-negative breast tumor. JCO Precis. Oncol. 1, 1–6 (2017).
Yang, J. J. et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin. Cancer Res. 20, 1383–1392 (2014).
Hijiya, N. et al. Phosphorylation status of epidermal growth factor receptor is closely associated with responsiveness to gefitinib in pulmonary adenocarcinoma. Hum. Pathol. 39, 316–323 (2008).
Li, S. et al. Phosphorylation of mTOR and S6RP predicts the efficacy of everolimus in patients with metastatic renal cell carcinoma. BMC Cancer 14, 376 (2014).
Hung, M. C., Wang, W. P. & Chi, Y. H. AKT phosphorylation as a predictive biomarker for PI3K/mTOR dual inhibition-induced proteolytic cleavage of mTOR companion proteins in small cell lung cancer. Cell Biosci. 12, 122 (2022).
Giuliani, R. et al. Phosphorylated HER-2 tyrosine kinase and Her-2/neu gene amplification as predictive factors of response to trastuzumab in patients with HER-2 overexpressing metastatic breast cancer (MBC). Eur. J. Cancer 43, 725–735 (2007).
Thor, A. D. et al. Activation (tyrosine phosphorylation) of ErbB-2 (HER-2/neu): a study of incidence and correlation with outcome in breast cancer. J. Clin. Oncol. 18, 3230–3239 (2000).
Sanchez, I. M. et al. In vivo ERK1/2 reporter predictively models response and resistance to combined BRAF and MEK inhibitors in melanoma. Mol. Cancer Ther. 18, 1637–1648 (2019).
Kato, S. et al. Functional measurement of mitogen-activated protein kinase pathway activation predicts responsiveness of RAS-mutant cancers to MEK inhibitors. Eur. J. Cancer 149, 184–192 (2021).
Liang, Y. et al. Phosphorylated ERK is a potential prognostic biomarker for Sorafenib response in hepatocellular carcinoma. Cancer Med. 6, 2787–2795 (2017).
Serrels, A. et al. Identification of potential biomarkers for measuring inhibition of Src kinase activity in colon cancer cells following treatment with dasatinib. Mol. Cancer Ther. 5, 3014–3022 (2006).
Bardia, A. et al. Biomarker analyses in the phase III ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. Ann. Oncol. 32, 1148–1156 (2021).
Johnson, D. B. et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 7, 10582 (2016).
Teng, P.-N et al. ProteoMixture: a cell type deconvolution tool for bulk tissue proteomic data. iScience https://doi.org/10.1016/j.isci.2024.109198 (2024).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Fisher, N. C. et al. Biological misinterpretation of transcriptional signatures in tumor samples can unknowingly undermine mechanistic understanding and faithful alignment with preclinical data. Clin. Cancer Res. 28, 4056–4069 (2022).
Eckert, M. A. et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature https://doi.org/10.1038/s41586-019-1173-8 (2019).
Liu, Z. et al. Suboptimal cytoreduction in ovarian carcinoma is associated with molecular pathways characteristic of increased stromal activation. Gynecol. Oncol. 139, 394–400 (2015).
Zhang, Q., Wang, C. & Cliby, W. A. Cancer-associated stroma significantly contributes to the mesenchymal subtype signature of serous ovarian cancer. Gynecol. Oncol. 152, 368–374 (2019).
Neesse, A. et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut 68, 159–171 (2019).
Maller, O. et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat. Mater. 20, 548–559 (2021).
Wulfkuhle, J. D. et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J. Proteome Res. 7, 1508–1517 (2008).
Hunt, A. L. et al. Integration of multi-omic data in a Molecular Tumor Board reveals EGFR-associated ALK-inhibitor resistance in a patient with inflammatory myofibroblastic cancer. Oncologist https://doi.org/10.1093/oncolo/oyad129 (2023).
Gallagher, R. I. et al. Protein signaling and drug target activation signatures to guide therapy prioritization: therapeutic resistance and sensitivity in the I-SPY 2 Trial. Cell Rep. Med. 4, 101312 (2023).
Clark, A. S. et al. Neoadjuvant T-DM1/pertuzumab and paclitaxel/trastuzumab/pertuzumab for HER2(+) breast cancer in the adaptively randomized I-SPY2 trial. Nat. Commun. 12, 6428 (2021).
Bostner, J. et al. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast Cancer Res. Treat. 137, 397–406 (2013).
Randall, J. et al. Quantitative proteomic analysis of HER2 protein expression in PDAC tumors. Clin. Proteom. 21, 24 (2024).
National Cancer Institute, D. o. C. C. a. P. S. Recent Trends in SEER Age-Adjusted Incidence Rates, 2000-2021. Surveillance Research Program, Surveillance Systems Branch. (2022).
Chen, E. Y., Raghunathan, V. & Prasad, V. An overview of cancer drugs approved by the US Food and Drug Administration based on the surrogate end point of response rate. JAMA Intern. Med. 179, 915–921 (2019).
Baldelli, E. et al. in Molecular Profiling: Methods and Protocols (ed. Espina, V.) 149–169 (Springer, New York, 2017).
Beaubier, N. et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 10, 2384–2396 (2019).
Finkle, J. D. et al. Validation of a liquid biopsy assay with molecular and clinical profiling of circulating tumor DNA. NPJ Precis. Oncol. 5, 63 (2021).
Spitzer, M., Wildenhain, J., Rappsilber, J. & Tyers, M. BoxPlotR: a web tool for generation of box plots. Nat. Methods 11, 121–122 (2014).
Acknowledgements
This work was supported, in part, by a generous contribution from the Win Sheridan family funds, and by funds from the Inova Health Foundation and the ExxonMobil Spouse’s Club. The authors would like to acknowledge Dr. Paulette Mhawech-Fauceglia for histopathology image analysis support and Dr. Kelly Conrads for critically reviewing the manuscript. Disclaimer: The views expressed in this article are those of the author(s) and do not necessarily reflect the official policy or position of the Uniformed Services University of the Health Sciences (USUHS), Department of the Navy, Department of the Air Force, Department of the Army, Department of Defense, or the United States Government. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Contributions
Study concept and design: T.L.C., T.P.C., J.R., E.F.P. Data acquisition, analysis, and interpretation: A.L.H., M.M.M., K.N., J.R., A.N., J.D., B.C., C.M., S.M., M.S., L.J., W.S., T.A., N.W.B., G.L.M., J.D., A.B., E.F.P., T.P.C., T.L.C. Writing (original draft): A.L.H. Writing (review and editing): A.L.H., J.R., E.F.P., T.P.C., T.L.C. All authors gave final approval of the completed work and are accountable for accuracy and integrity.
Corresponding authors
Ethics declarations
Competing interests
T.P.C. is a ThermoFisher Scientific, Inc. SAB member and receives research funding from AbbVie. E.F.P. is a SAB member and consultant to Ignite Proteomics, Inc., and receives research funding from Genentech, Pfizer, Mirati, Springworks Therapeutics, Deciphera, AbbVie, and is a co-inventor of the RPPA Technology described herein, and related HER2 biomarker patents and receives royalties on the related license agreements. K.N. is an independent consultant and receives funding from Catalyst Medical Media, Clinical Care Options, Global Healthcare Marketing & Communications, Health and Wellness Partners, Interactive Forums, MphaR, and PharMecha. J.D., B.C., and C.M. are full-time employees of Ignite Proteomics, Inc.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hunt, A.L., Randall, J., Mansukhani, M.M. et al. Real-time functional proteomics enhances therapeutic targeting in precision oncology molecular tumor boards. npj Precis. Onc. 9, 111 (2025). https://doi.org/10.1038/s41698-025-00868-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00868-y
