
Abstract
Aptamers are short single-stranded DNA or RNA oligonucleotides that are selected in vitro by their affinity and specificity for the target. Binding is a consequence of the particular tertiary structure that they are able to acquire, depending on their sequence. Parasites of the genus Leishmania belongs to the lower eukaryote order Kinetoplastida that causes leishmaniosis in man and animals. Histone genes in Leishmania are of considerable interest because these flagellates do not condense their chromatin during mitosis. Thus, the study of the structural features of histones has been considered of particular interest and, as a result, in recent years a great number of histone genes have been characterized in trypanosomatids. Histones are extremely conserved proteins, reflecting their apparent universality of function. Sequence similarity of kinetoplastid core histones those of higher eukaryotes is found predominantly in the globular region with high sequence divergences in the N- and in the C-terminal domains. These divergences indicate that they may be potential diagnostic and/or therapeutics targets. We have successfully isolated a pool of DNA sequences, named SELH2A, which specifically binds to Leishmania infantum H2A. When tested in an enzyme-linked oligonucleotide assay, slot blot and Western blot analysis, the aptamer pool exhibited specificity in its ability to bind only to H2A antigen but not to other proteins from L. infantum including other histones. Thus, it appears that this novel anti-H2A aptamer population may be of potential application as a diagnostic system for leishmaniosis.
Similar content being viewed by others

Main
Aptamers are single-stranded oligonucleotides that are selected from combinatorial libraries by systemic evolution of ligands using exponential enrichment (SELEX) technology, and are capable of selectively binding target molecules with high affinity.1, 2 These single-stranded (ss) nucleic acid molecules have highly defined tertiary structures that are formed owing to the ability of short sequences to fold into unique three-dimensional structures in the presence of a ligand, and they bind to their targets with high affinity and specificity. It has been shown that aptamers form stable and specific complexes with a range of different targets, including small molecules such as amino acids and highly complex proteins and whole viruses.3, 4, 5, 6, 7, 8, 9, 10 These specialized molecules are analogs to antibodies in specificity and affinity, but have the potential advantage of being reproduced by chemical synthesis and be more easily labeled with fluorescent or other reporters during their synthesis. Consequently, they are considered potential therapeutic agents and also thought to rival antibodies in immunoassay-like analyses. On comparing aptamers with proteins that bind to the same targets have shown that both nucleic acids and proteins use similar strategies for the formation of well-defined binding patterns.11, 12 Structural studies with aptamer-target complexes have demonstrated insights into molecular diversity associated with nucleic acid architecture and molecular recognition.13 Aptamers frequently form complexes that have dissociation constants in the nanomolar range and can clearly distinguish between even closely related protein targets.14, 15
The procedure for obtaining aptamers requires the incubation of a target with a library of DNA sequences, typically ranging from 1014 to 1018 molecules in complexity. Target–DNA complexes are isolated, the DNA is amplified and the process is repeated until the desired sample is enriched with sequences that display high affinity and specificity for the target of interest. Starting from a library of vast diversity, it is often possible to identify aptamers with nanomolar or subnanomolar affinities. Aptamers offer advantages over antibody-based affinity because of the nature of DNA/RNA, which provides increased stability, easy regeneration (PCR or chemical synthesis) and simple modifications for detection and neutralization. On the contrary, antibodies in general are stable only under physiological conditions and any modifications often affect the affinity for targets.
Leishmaniasis is endemic in 88 countries on four continents. The causative agent is a parasitic protozoa of the genus Leishmania, transmitted to humans by sandflies. Over 20 species and subspecies infect humans, each causing a different spectrum of symptoms. These range from simple, self-healing skin ulcers (eg owing to infection with L. major), to severe, life-threatening disease (eg visceral leishmaniasis caused by L. donovani). Humans are infected via the bite of sandflies (subfamily phlebotominae) that breed in forest areas, caves, or the burrows of small rodents. Wild and domesticated animals and humans themselves can act as a reservoir of infection. Most forms of leishmaniasis are originally infections of small mammals (‘reservoir hosts’), which play a major role in the epidemiology of the disease. Sandflies become infected by ingesting blood from infected reservoir hosts or from infected people.
Leishmania parasites possess a digenetic life cycle with two discrete morphological phases: the promastigote, which develops extracelullarly within the gut of the insect vector, and the amastigote, which is specialized to survive within the macrophage phagolysosome of vertebrate host. These parasites, like other related kinetoplastid protozoa, are placed in the most primitive branch of the eukaryote evolution and possess very peculiar features of gene expression and organization. Among them, the organization of the nuclear genome differs from that of higher eukaryotes. The nuclear envelope persists during cell division and chromosomes are not visualized at any phase of the cell cycle. Although chromatin is organized in nucleosomes, higher-order structures of 30 nm fibers are not observed. Despite the fact that histones are among the most highly conserved protein along the evolutionary scale, histones of trypanosomatids have accumulated substantial sequence differences, mainly at the N- and C-terminal region.16 In fact, the specificity of the humoral response ellicited against parasite histones during Leishmania infection is because of the localization of the main antigenic determinants in those divergent regions.17 This helped to develop a recombinant multiepitope protein containing the antigenic determinants of the H2A histone that represent a valuable tool for the serodiagnosis of canine visceral leishmaniosis.18
Here, we report the selection of a ssDNA aptamer population, designated as SELH2A, that specifically recognizes L. infantum H2A antigen, and that can be used as a screening tool for detection and identification of H2A antigen with detection limits as low as 50 ng of protein using optical detection. In this paper, we detail the application of these aptamers in enzyme-linked oligonucleotide assay (ELONA), slot blot and Western blot assays. In addition, we have identified the regions in H2A that are recognized by SELH2A aptamer population to map the H2A–aptamer interaction. Thus, our findings futher support that aptamers may represent a new class of reagents that might be used in the detection and diagnostic platforms that were previously by antibodies.
Materials and methods
Materials
Synthetic random DNA and non-labeled primers were obtained from Metabion (Martinsried, Germany) and digoxigenin-labeled primers were purchased from TIB MOLBIOL (Berlin, Germany). Bovine serum albumin (BSA) was purchased from Sigma. Dulbeco's modified Eagle's medium (DMEM) and PBS were obtained from GibcoBRL.
Expression and Purification of Recombinant H2A
L. infantum H2A protein was previously cloned in the pQe30 expression vector and the recombinant H2A protein was purified by affinity chromatography on Ni-NTA resin columns as described.19 Briefly, the cells expressing recombinant H2A were harvested and re-suspended in sonication buffer (8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-HCl, pH 8.0) at 2–5 volumes g−1 of wet weight. After binding to a Ni-nitroacetic acid column recombinant proteins were gradually refolded on the affinity column as described.20 Afterwards, recombinant H2A was eluted with 0.3 M imidazol, dialyzed against PBS at 4°C and lyophilized.
In Vitro Selection Procedure
Iterative rounds of selection and amplification of ssDNA aptamers were performed as described previously by Morris et al.21 In brief, ssDNA oligonucleotides, designated as RND40, contained a central randomized region of 40 nucleotides flanked by two conserved 18-nucleotides regions in each end (5′-GCGGATGAAGACTGGTCT-40N-GTTGCTCGTATTTAGGGC-3′). The ssDNA library was denatured at 90°C for 10 min and then cooled on ice for 10 min. For the initial SELEX round, 2 nmol of ssDNA were mixed with 2 μg of recombinant His-tagged H2A in 200 μl of selection buffer (20 mM Tris-HCl, pH 7.4, 1 mM MgCl2, 150 mM NaCl, 5 mM KCl, 0.2% BSA) and incubated at 37°C for 30 min. The bound aptamer–H2A complexes were purified by adding 20 μl of Ni-NTA superflow (Qiagen) for 1 h at 4°C. After washing three times with 1 ml of selection buffer, the ssDNA-protein complexes were re-suspended in 20 μl of distilled H2O and amplified by PCR using the primers named F3 (5′-GCGGATGAAGACTGGTGT-3′) and R3 (5′-GTTGCTCGTATTTAGGGC-3′) under the conditions of 1 μM/primer, 250 μM dNTPs, in a final volume of 200 μl at 56°C for 25 cycles. PCR product was ethanol precipitated. After round 3, the pool, named SELH2A, was amplified and labeled by PCR using digoxigenin-labeled primer F3 and non-labeled primer R3.
Enzyme-Linked OligoNucleotide Assay
Recombinant L. infantum H2A protein was diluted to 5 μg/ml (330 nM) in selection buffer and 200 μl of the solution (66 pmol/well) were incubated in a 96-well microtiter plate (Costar) overnight at 4°C to allow plate binding and, then, washed four times in selection buffer. Afterwards, 200 μl of digoxigenin-labeled SELH2A aptamer population or digoxigenin-labeled RND40 library in selection buffer diluted at different concentrations (0.2–100 nM), previously denatured for 10 min at 95°C and cooled for 10 min on ice, were added to each well. The plate was incubated at 37°C for 30 min after which individual wells were washed four times with selection buffer to remove unbound ssDNA. Afterwards, 200 μl of a 1:1000 dilution of anti-digoxigenin-POD antibody was added to the individual wells. Following a 30-min incubation at 37°C on a shaking platform, the plates were washed four times and developed using ABTS solution (Boehringer–Mannheim) according to the manufacturer’s instruction. OD405 values were determined using a microplate reader from TECAN.
In other set of experiments, recombinant H2A protein was diluted to the required concentrations (1.65–660 nM) in selection buffer and 200 μl of each solution were incubated in a 96-well microtiter plate (Costar) overnight at 4°C and washed four times. Digoxigenin-labeled SELH2A aptamer population was diluted in selection buffer at 20 nM concentration and then denatured for 10 min at 95°C and cooled for 10 min on ice. Next, 200 μl of aptamers were added to each well and the plate was incubated at 37°C for 30 min and individual wells were washed three times, anti-digoxigenin-POD antibody was added and developed using ABTS solution (Boehringer–Mannheim) as above.
Similar experiments were performed coating the wells with 2 μg of other Leishmania proteins (H2B, H3, LIP0, LiP2a and LiP2b) to assess the specificity of the aptamers.
Slot Blot Analysis with Aptamers
Several amounts of recombinant L. infantum H2A protein (1.65–33 pmol) and 7.5 pmol of BSA were transferred onto nitrocellulose membranes under vacuum. Filter was washed three times in PBS-T for 10 min and then blocked with 5% milk in PBS-T for 1 h at room temperature. Afterwards, membranes were incubated with digoxigenin-labeled SELH2A aptamer population at 100, 200 and 400 nM in selection buffer for 1 h at room temperature with gentle rocking. After the designated time, membranes were washed with selection buffer three times and probed with anti-digoxigenin antibody conjugated with horseradish peroxidase (POD) (Roche) diluted 1/1000, for 1 h at room temperature. Excess enzyme was removed by three subsequent washes with selection buffer. Finally, the membranes were developed with enhanced chemiluminescence kits (Amersham Biosciences) and exposed to hyperfilm. BSA was used as negative control.
Binding Kinetics Studies
To determine the kinetics of binding of SELH2A to H2A, we performed ELONA experiments as above in which recombinant H2A protein (66 pmol/well) was incubated with digoxigenin-labeled SELH2A aptamer population (4 pmol/well) at 37°C for several incubation times after which individual wells were washed four times with selection buffer to remove unbound ssDNA. Afterwards, anti-digoxigenin-POD antibody was added and developed using ABTS solution (Boehringer–Mannheim) as above.
Immunoblotting
Promastigotes from L. infantum were grown in DMEM containing 10% of fetal bovine serum at 27°C. Total protein extracts were obtained from promastigotes of L. infantum that were pelleted and lysed in lysis buffer (125 mM Tris-HCl, pH 6.8, 4% SDS, 15 mM EDTA, 20% glycerol, 10% β-mercaptoethanol and 0.008% bromophenol blue). Nuclear and cytosolic fractions were separated according to Schreiber et al.22 Briefly, 2 × 107 promastigotes were pelleted, washed two times in ice-cold PBS, re-suspended in 400 μl of prechilled buffer A (10 mM HEPES, pH 7.5, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 0.5 mM PMSF) and incubated 15 min on ice. After incubation NP40 was added (0.6% final), and cells were lysed by vigorous vortexing for 10 s and immediately pelleted in a microfuge (13 000 g). The supernatant (cytosolic fraction) was mixed 1:1 in 2 × Laemmli’s buffer. The pelleted nuclei were re-suspended in 1 × Laemmli’s buffer. Afterwards, membranes were incubated with digoxigenin-labeled SELH2A aptamer population at 40 nM in selection buffer for 1 h at room temperature with gentle rocking and then washed with selection buffer three times and probed with anti-digoxigenin-POD antibody diluted 1/1000, for 1 h at room temperature. Finally, after three subsequent washes with selection buffer, the membranes were developed with enhanced chemiluminescence kits (Amersham Biosciences) and exposed to hyperfilm.
Mapping of the H2A–SELH2A Interaction
Nine peptides corresponding to overlapped sequences of L. infantum H2A protein (10 μg/well) were incubated in a 96-well microtiter plate (Costar) overnight at 4°C and, then, washed four times. Afterwards, digoxigenin-labeled SELH2A aptamer population at 0.5 μg/ml was denatured for 10 min at 95°C, cooled for 10 min on ice and 200 μl of the solution was added to each well. Next, plate was incubated at 37°C for 30 min, washed four times, and anti-digoxigenin-POD antibody was added at 37°C for 30 min and developed using ABTS solution (Boehringer–Mannheim) as above. Absorbance at 405 nm was determined using a microplate reader from TECAN. Peptides were synthesized by the simultaneous multiple solid-phase synthetic method using a polyamine resin and FMOC chemistry.23 Purity was checked by amino-acid analysis and HPLC.
Statistical Analysis
Data are presented as mean value±s.e.m. from three to six independent measurements in separate experiments and analyzed using GraphPad Prism v4 (San Diego, CA, USA). The different statistical analyses used are indicated in the text and in the figure legends.
Results
Selection of Anti-H2A Aptamers from Combinatorial Library
DNA aptamers specific for L. infantum H2A protein were selected from a library of ssDNA with a 40-nucleotide random region flanked by 18-nt 5′ and 3′ fixed regions for PCR amplification. The initial library theoretically contained 1014–1016 different sequences. Upon mixing with H2A, oligomers from the library may bind to the target. During this process a bound candidate or candidates of the pool may be specific for the target and can be amplified with primers F3 and R3. The number of PCR cycles was also optimized to avoid overamplification. The stringency of the selection was controlled by adjusting the antigen concentrations, buffer conditions and the washings. ssDNA sequences exhibiting affinity to the target were captured by agarose-conjugated Ni beads. The affinity selection was stopped after third round, and the selected population, named SELH2A, was amplified by PCR using digoxigenin-labeled primer F3 and non-labeled primer R3.
The H2A-binding properties of the starting RND40 and final SELH2A pools were compared in an ELONA, which provides a rapid assessment of the relative binding capabilities of the two populations. As it can be observed in Figure 1, the SELH2A population displayed increased binding to H2A protein compared with the starting pool. These data clearly demonstrate that SELH2A is enriched in ssDNA sequences that recognize H2A protein relative to RND40. In view of these results, we decided to characterize different aspects of the SELH2A population to determine whether or not this aptamer population could be used in a Leishmania detection system.

Relative H2A binding of SELH2A aptamer population and RND40 initial pool. SELEX pool from round 3 and the starting round were assayed for binding to Leishmania H2A protein. Binding studies were performed by ELONA as described in ‘Materials and methods’. Recombinant protein H2A was plated at 66 pmol/well and incubated with 200 μl of digoxigenin-labeled SELH2A aptamer population or digoxigenin-labeled RND40 library at 5, 10, 20 and 100 nM.
Binding Affinity of SELH2A Aptamer Population to L. infantum H2A by ELONA
To study the affinity of SELH2A aptamers for H2A protein, we performed an ELONA assay in which the 96-well plates were coated with recombinant H2A protein and several amounts of digoxigenin-labeled SELH2A was tested as described in Materials and methods. Data were analyzed using nonlinear regression showing that they respond to a one-site binding curve with an equation y=(x × Bmax)/(x+KD), where Bmax is the maximal binding and KD is the concentration of ligand required to reach half-maximal binding. Data presented in Figure 2a indicate that SELH2A population is able to detect recombinant H2A protein in a concentration-dependent manner with a KD=2.065±0.652 nM, significantly lower than that for RND40 (KD=45.24±18.37 nM). A more exhaustive analysis of the ELONA data represented in Figure 2a (inset) showed that concentrations greater than 1 pmol of SELH2A was able to detect from 66 pmol of recombinant H2A protein in a significant manner (P<0.01; ANOVA followed by Dunnett's test).

Binding capability of the SELH2A aptamer population for L. infantum H2A proteins assessed by ELONA. The assays were performed as described in ‘Materials and methods’. (a) Recombinant protein H2A and BSA were plated at 66 pmol/well and incubated with several concentrations (0.2–100 nM) of digoxigenin-labeled SELH2A aptamer population. (b) Recombinant H2A protein was plated to several concentrations (0.33–132 pmol/well) and incubated with 4 pmol/well of digoxigenin-labeled SELH2A aptamer population. Finally, anti-digoxigenin-POD antibodies were added plate was revealed with ABTS® solution at 405 nm. Inset in (a) and (b) show the data as a bar graphs. All the experiments were made in triplicate and average of four different experiments is shown in the figure. Statistical significance between each concentration of SELH2A population (a) or H2A protein (b) and the value obtained for the blank (b, P<0.01).
To determine the sensitivity of the selected polyclonal DNA aptamer population SELH2A we performed an analysis in which 96-well plates were coated with quantities ranging from 0.33 to 132 pmol of recombinant H2A or 30 pmol of BSA protein and incubated in the presence of digoxigenin-labeled SELH2A as described in Materials and methods. The saturation curve (Figure 2b), fitted as above, showed that SELH2A population is able to detect recombinant H2A protein in a concentration-dependent manner. As seen in Figure 2b, it is further confirmed that the protein affinity to the aptamer population is directly proportional to the quantity of H2A until 33 pmol of protein (R2=0.9313, P<0.0001; linear regression). A more detailed analysis of the data (Figure 2b, inset) indicates that 4 pmol of SELH2A aptamer population is able to detect from 16.5 pmol of H2A protein in a significant manner (P<0.01; ANOVA followed by Dunnett's test).
Recognition of L. infantum H2A Protein by SELH2A Population
In view of the above results, we decided to study the binding of SELH2A population to the target using an alternative technique. Thus, we performed slot blot experiments in which several amounts of recombinant H2A protein and BSA were transferred onto nitrocellulose membranes and the immobilized proteins were probed with three different concentrations of digoxigenin-labeled SELH2A aptamer population as described in Materials and methods. As shown in Figure 3, SELH2A population showed strong affinity to recombinant H2A protein but did not show any affinity to negative control BSA, thereby indicating their specificity towards the H2A antigen. In these type of experiments, as few as 1.65 pmol of H2A was detected after incubation of the nitrocellulose membrane with 100 nM SELH2A. All the above results confirm that SELH2A recognizes recombinant H2A protein with high affinity but not BSA.

Slot blot analysis of the specificity of the SELH2A aptamer population. BSA and H2A at the concentration indicated in the figure were fixed under vacuum to a nitrocellulose membrane. Membranes were blotted with digoxigenin-labeled SELH2A aptamer population at several concentrations. Afterwards, the membrane was probed with anti-digoxigenin-POD antibodies, developed with enhanced chemiluminescence's kits and exposed to hyperfilm. The experiment shown is representative of at least three different experiments.
Binding Kinetics of SELH2A Aptamer Population to L. infantum H2A by ELONA
The fact that SELH2A concentrations between 2 and 20 pmol/well do not show increased binding to H2A could be indicating that, after a 30-min incubation, only a small population of aptamers is able to bind to H2A. In view of this result, we decided to study the binding kinetics of SELH2A in an ELONA assay as described in Materials and methods. Thus, 66 pmol of H2A was incubated with 4 pmol of digoxigenin-labeled SELH2A for 5, 10, 20, 40, 60, 80 and 100 min and absorbance at 405 nm was determined after incubation with anti-digoxigenin-POD antibody. Results clearly indicate that SELH2A binds to H2A protein in a time-dependent manner (r=1; P<0.0001; Spearman correlation) reaching a plateau after 60 min of incubation (Figure 4). However, anything above 5 min is statistically significant for control and BSA. In view of these results, we consider that a 30-min incubation is time enough to maximize aptamer binding to the target.

Binding kinetics of SELH2A aptamer population to L. infantum H2A by ELONA. Recombinant protein H2A and BSA were plated at 66 pmol/well and incubated with 20 pmol/well of digoxigenin-labeled SELH2A aptamer population for 5, 10, 20, 40, 60, 80 and 100 min. Afterwards, anti-digoxigenin-POD antibody were added and plate was revealed with ABTS® solution at 405 nm. All the experiments were made in triplicate and average of four different experiments is shown in the figure.
Binding Specificity of SELH2A Aptamers to L. infantum Proteins
To study the specificity of SELH2A population to other proteins from L. infantum, we tested the binding affinity of these aptamers against H2B, H3, LIP0, LiP2a and LiP2b by ELONA. As can be observed in Figure 5a, the signal output is almost 8000 for H2A and 4000 for H3. The signal for the other proteins is around 2000, similar to that for negative control. From these results it can be concluded that SELH2A specifically recognizes both H2A and H3 (P<0.01; ANOVA followed by Tukey's test) but H3 with a significant lower signal than H2A (P<0.001). This, most likely, is owing to some structural homology between H2A and H3. In contrast, the related protein H2B and the ribosomal proteins LIP0, LiP2a and LiP2b have to have different structural characteristics. We have also made some ELONA experiments using histones purified from cow cells and the results indicated that SELH2A seems to recognize H2A from this source although with a lower affinity (data not shown).

Specificity of the SELH2A aptamer population assessed by ELONA and Western blot assays. (a) Recombinant proteins H2A, H2B, H3, LIP0, LiP2a, LiP2b and BSA were plated at 2 μg/well (≈132 pmol/well) and incubated with 200 μl of 20 nM of digoxigenin-labeled SELH2A aptamer population. Afterwards, anti-digoxigenin-POD antibody was added and the plates were developed using ABTS solution. Abs 405 nm values were determined using a microplate reader from TECAN. All the experiments were made in triplicate and average of four different experiments is shown in the figure. Statistical significance with respect to the blank (a, P<0.001). Statistical significance between H2A and H3 (***P<0.001). (b) Thirty micrograms of total proteins (lane T) and nuclear proteins (lane N) from L. infantum (left panel or human cells (right panel) and 1 μg of recombinant H2A (lane R) were separated on SDS-15% PAGE gels and transferred to nitrocellulose membranes. Membranes were incubated with 1 μg/ml (40 nM) of the digoxigenin-labeled SELH2A aptamer population and probed with anti-digoxigenin-POD antibody. Finally, the membranes were developed with enhanced chemiluminescence's kits and exposed to hyperfilm. The sizes (in kDa) of the molecular size markers are indicated on the left. All the experiments were made in triplicate. Picture shows a representative experiment.
To verify the above results, we also performed parallel Western blot assays in which total protein and nuclear fraction from promastigotes of L. infantum or human cells and recombinant H2A protein were immobilized on PVDF membranes and then probed with digoxigenin-labeled SELH2A aptamer population and detected with anti-digoxigenin-POD antibody. As shown in Figure 5b, the selected-aptamers specifically recognize endogenous H2A in lanes corresponding to both total proteins and nuclear fraction from L. infantum promastigotes (left panel) but, however, this band does not appear in nuclear fraction and, with a very low intensity, in total lysate from human cells (right panel). In the same experiment, we included recombinant H2A protein as a positive control. The intensity of this band can be used as a reference.
Mapping of the H2A–SELH2A Interaction
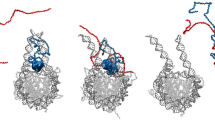
To determine the H2A regions that are recognized with higher affinity by SELH2A population, we performed ELONA assays using nine peptides corresponding to overlapped sequences of H2A protein as antigen (Figure 6a). As shown in Figure 6b, SELH2A recognized peptides 5 and 8 with very high affinity (P<0.01; ANOVA followed by Dunnett's test). Moreover, peptide 2 was also recognized but with a significantly lower affinity (P<0.05). Next, we decided to locate the peptides on the tertiary structure of H2A. Figure 6c shows that peptides 5 and 8 are positioned forming a pocket region corresponding to a side of the protein. This region is accessible when H2A is forming part of the nucleosome (Figure 6d).

Mapping of the H2A-SELH2A interaction. (a) Sequence of the peptides corresponding to overlapped sequences of H2A protein. (b) ELONA assay showing the binding ability of SELH2A to the different peptides. All the experiments were made in triplicate and average of four different experiments is shown in the figure (b, P<0.01; c, P<0.05). (c) Localization of the peptides recognized by SELH2A on the tertiary structure of H2A. (d) Localization of the peptides recognized by SELH2A on the nucleosome structure.
Discussion
In this study, L. infantum H2A antigen was selected as a target because this protein is considered an excellent candidate for Leishmania diagnosis24, 25 and therapy.19 Because it has been shown that aptamers are able to discriminate closely related molecules and also exhibit chiral discrimination of their targets15 this technology could be useful to select ssDNA aptamers with high affinity and specificity for H2A protein relative to other histones. It is reasonable to assume that DNA aptamers without further modifications could perform better in detection and diagnostic assays in which the aptamers may come in contact with different biological samples for a brief period of time. Although other types of detection and diagnostic platforms utilizing peptides and antibodies are available, DNA-based technology may be more suitable for the detection of Leishmania in environmental conditions in which this parasite is endemic.
Enzyme linked assays have provided a means of quickly evaluating the binding affinity. Drolet et al26 reported the first enzyme-linked aptamer assay (ELAA) consisting of a mixed ELISA/ELAA sandwich to detect human vascular endothelial growth factor on a microtiter plate. Since then, similar assays have been used with either labeled or immobilized nucleic acid molecules as capturing or detecting agents. Aptamer-conjugated immunoassays using other platforms to detect specific proteins were reported earlier.27, 28 The most remarkable applications were the demonstration of a fiber-optic microarray biosensor using aptamers29 and an aptamer-based quartz crystal biosensor.30 None of these reports utilized nucleic acid-based enzymatic assays as described, consisting in an aptamer sandwich assay with digoxigenin-labeled aptamers in a 96-well microtiter plate format. Very recently, in a displacement enzyme-linked aptamer assay using thrombin, Baldrich et al31 also demonstrated the superior performance of aptamers compared with antibodies.
In our study, we have selected an aptamer population, named SELH2A, which is able to recognize H2A with a very low background signal. Using aptamers in slot blot and Western blot analysis is another means to study the affinity and specificity to the target. We have tested the selected aptamer population SELH2A for its binding to H2A antigen by slot blot analysis with peroxidase-conjugated enzymatic assays. The aptamer population showed high specificity for H2A but no cross-reactivity to BSA. In addition, Western blot results clearly showed that SELH2A aptamers were able to identify native H2A in a total lysate containing all the parasite proteins with a very low background. However, we have observed that although SELH2A also can identify H2A in total lysates from human cells with very low sensitivity, it mainly recognizes non-specifically several other proteins. These data demonstrate that the apparent lack of specificity observed in ELONA experiments using histones purified from cow cells is related to non-specific reactivity but not to cross-reactivity with histones from different species. These clear data strongly suggest that SELH2A aptamer population might be used as a tool to detect H2A from Leishmania, as there are no commercial antibodies against this protein.
In summary, we have generated a population of oligonucleotide anti-ligands that bind with high specificity to L. infantum H2A antigen. These ssDNA molecules specifically recognize H2A antigen from L. infantum and do not bind to other proteins of the parasite. The current work demonstrates that aptamers can be used for detection of specific targets in an ELONA formats. It is important to point out that aptamers have great potential to circumvent limitations associated with antibodies. Although the antibody detection system is currently the gold standard for protein detection and identification, the data presented in this study strongly support the feasibility of the aptamer-based diagnostic system. In conclusion, it is reasonable to expect that the ELONA platform and Western blot generated by aptamers will pave the way for future detection systems in which antibodies have been previously used.
References
Tuerk C, Gold L . Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990;249:505–510.
Ellington AD, Szostak JW . Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 1992;355:850–852.
Famulok M . Oligonucleotide aptamers that recognize small molecules. Curr Opin Struct Biol 1999;9:324–329.
Mannironi C, Di Nardo A, Fruscoloni P, et al. In vitro selection of dopamine RNA ligands. Biochemistry 1997;36:9726–9734.
Nieuwlandt D, Wecker M, Gold L . In vitro selection of RNA ligands to substance P. Biochemistry 1995;34:5651–5659.
Green LS, Jellinek D, Jenison R, et al. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 1996;35:14413–14424.
Takeno H, Yamamoto S, Tanaka T, et al. Selection of an RNA molecule that specifically inhibits the protease activity of subtilisin. J Biochem (Tokyo) 1999;125:1115–1119.
Bock LC, Griffin LC, Latham JA, et al. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992;355:564–566.
Moreno M, Rincon E, Pineiro D, et al. Selection of aptamers against KMP-11 using colloidal gold during the SELEX process. Biochem Biophys Res Commun 2003;308:214–218.
Green LS, Jellinek D, Bell C, et al. Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem Biol 1995;2:683–695.
Hermann T, Patel DJ . Adaptive recognition by nucleic acid aptamers. Science 2000;287:820–825.
Marshall KA, Robertson MP, Ellington AD . A biopolymer by any other name would bind as well: a comparison of the ligand-binding pockets of nucleic acids and proteins. Structure 1997;5:729–734.
Gold L, Polisky B, Uhlenbeck O, et al. Diversity of oligonucleotide functions. Annu Rev Biochem 1995;64:763–797.
Uphoff KW, Bell SD, Ellington AD . In vitro selection of aptamers: the dearth of pure reason. Curr Opin Struct Biol 1996;6:281–288.
Jenison RD, Gill SC, Pardi A, et al. High-resolution molecular discrimination by RNA. Science 1994;263:1425–1429.
Galanti N, Galindo V, Sabaj V, et al. Histone genes in Trypanosomatids. Parasitology Today 1998;14:64–70.
Requena JM, Alonso C, Soto M . Evolutionarily conserved proteins as prominent immunogens during Leishmania infections. Parasitol Today 2000;16:246–250.
Soto M, Requena JM, Quijada L, et al. Multicomponent chimeric antigen for serodiagnosis of canine visceral leishmaniasis. J Clin Microbiol 1998;36:58–63.
Iborra S, Soto M, Carrion J, et al. Vaccination with a plasmid DNA cocktail encoding the nucleosomal histones of Leishmania confers protection against murine cutaneous leishmaniosis. Vaccine 2004;22:3865–3876.
Shi PY, Maizels N, Weiner AM . Recovery of soluble, active recombinant protein from inclusion bodies. Biotechniques 1997;23:1036–1038.
Morris KN, Jensen KB, Julin CM, et al. High affinity ligands from in vitro selection: complex targets. Proc Natl Acad Sci USA 1998;95:2902–2907.
Schreiber E, Matthias P, Muller MM, et al. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 1989;17:6419.
Houghten RA . General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc Natl Acad Sci USA 1985;82:5131–5135.
Soto M, Requena JM, Gomez LC, et al. Molecular characterization of a Leishmania donovani infantum antigen identified as histone H2A. Eur J Biochem 1992;205:211–216.
Soto M, Requena JM, Quijada L, et al. Mapping of the linear antigenic determinants from the Leishmania infantum histone H2A recognized by sera from dogs with leishmaniasis. Immunol Lett 1995;48:209–214.
Drolet DW, Moon-McDermott L, Romig TS . An enzyme-linked oligonucleotide assay. Nat Biotechnol 1996;14:1021–1025.
Rye PD, Nustad K . Immunomagnetic DNA aptamer assay. Biotechniques 2001;30:290–292, 294–5.
Yang X, Li X, Prow TW, et al. Immunofluorescence assay and flow-cytometry selection of bead-bound aptamers. Nucleic Acids Res 2003;31:e54.
Lee M, Walt DR . A fiber-optic microarray biosensor using aptamers as receptors. Anal Biochem 2000;282:142–146.
Liss M, Petersen B, Wolf H, et al. An aptamer-based quartz crystal protein biosensor. Anal Chem 2002;74:4488–4495.
Baldrich E, Acero JL, Reekmans G, et al. Displacement enzyme linked aptamer assay. Anal Chem 2005;77:4774–4784.
Acknowledgements
ER is a fellow from Comunidad Autónoma de Madrid (FINNOVA program), DP is a fellow from Fondo de Investigaciones Sanitarias (FIS 03/0469), MEM is a researcher from FIBio-HRC supported by Consejeria de Sanidad y Consumo (CAM) and VMG is supported by a research contract from FIS. This work was supported by Grant FIS05/0453 from Ministerio de Sanidad y Consumo (Spain).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramos, E., Piñeiro, D., Soto, M. et al. A DNA aptamer population specifically detects Leishmania infantum H2A antigen. Lab Invest 87, 409–416 (2007). https://doi.org/10.1038/labinvest.3700535
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/labinvest.3700535
Keywords
This article is cited by
-
Targeting the polyadenylation factor EhCFIm25 with RNA aptamers controls survival in Entamoeba histolytica
Scientific Reports (2018)
-
Inhibition of Influenza Virus Replication by DNA Aptamers Targeting a Cellular Component of Translation Initiation
Molecular Therapy - Nucleic Acids (2016)
-
Characterization of MNK1b DNA Aptamers That Inhibit Proliferation in MDA-MB231 Breast Cancer Cells
Molecular Therapy - Nucleic Acids (2016)
-
Development of a Fluorescent Enzyme-Linked DNA Aptamer-Magnetic Bead Sandwich Assay and Portable Fluorometer for Sensitive and Rapid Leishmania Detection in Sandflies
Journal of Fluorescence (2014)
