
Abstract
Non-Hodgkin’s lymphomas uncommonly present as bone lesions. Most of these tumors are diffuse large B-cell lymphomas. Anaplastic large cell lymphoma (ALCL) presented as bone lesions is exceedingly rare. In this study, we describe six cases of ALCL that presented as solitary or multiple bone lesions. The average patient age was 33 years (range, 4 to 63 years) and the male to female ratio was 2:1. Fever and localized bone pain were the most frequent presenting symptoms. Radiologic examinations revealed osteolytic lesions in all cases, with three (50%) being multiple lesions and five (83%) involving the axial bones. All patients were initially assessed to have only bone involvement. Staging studies revealed mild cervical lymphadenopathy in one patient and no evidence of extraskeletal disease in the other five patients. Histologically, there was diffuse infiltration of one or more bones by large pleomorphic lymphoma cells. Immunohistochemical studies showed all six neoplasms were positive for CD30, EMA, and granzyme B. One case was of T-cell lineage, positive for CD3. One case was positive for the T-cell-associated antigen CD4. The remaining four cases were of null-cell type. In-situ hybridization for EBV was performed in five cases; all were negative. Despite the relatively low International Prognostic Index (IPI) of these patients (mean, 1.67; range, 1 to 3), the overall prognosis was relatively poor: three of six died of disease within 2 years of diagnosis, and two of six were alive with evidence of disease (follow-up, 6 mo to 2 years). Thus, compared to their nodal counterparts, ALCLs that present as bone lesions are distinguished by their uniform expression of EMA and granzyme B, and a relatively poor clinical outcome. Our results also suggest that ALK-1 expression in this clinical setting is not a favorable prognostic indicator.
Similar content being viewed by others
INTRODUCTION
Anaplastic large cell lymphoma (ALCL) is a type of non-Hodgkin’s lymphoma that is most often composed of large pleomorphic lymphoid cells that express the CD30 antigen and have a propensity to involve lymph node sinuses (1). Although most of these lymphomas are of T/null lineage, some reported cases have been of B-cell or null-cell lineage. Subsequently, the definition of ALCL has been modified to incorporate additional clinical and molecular data (2, 3). A subset of T-cell or null-cell ALCL carries a characteristic cytogenetic abnormality, the t(2;5)(p23;q35), which results in ALK-1 protein expression and has been found to be associated with a young patient age and good prognosis (4, 5, 6). In contrast, most CD30-positive large B-cell lymphomas with anaplastic cytologic features do not carry the t(2;5)(p23;q35), are negative for ALK-1, and clinically behave similar to diffuse large B-cell lymphomas (7, 8). Thus, the diagnosis of ALCL is currently restricted to those of T/null-cell type in the Revised European and American Lymphoma Classification (REAL) and the recently proposed World Health Organization (WHO) Classification of Lymphomas (9, 10), and ALK-1 expression is regarded as an important prognostic marker for ALCL. It also has been suggested that the clinical behavior and pathogenesis of ALCL may differ according to the anatomic sites involved. One example is primary cutaneous ALCL, which differs from their nodal counterparts in that they lack ALK-1 expression and the t(2;5)(p23;q35) and carry a favorable prognosis (11).
Primary bone lymphomas are uncommon, and most of these tumors are diffuse large B-cell lymphomas (12). Although ALCL most commonly involves lymph nodes, a few case studies in the literature have described ALCL presented primarily as one or more bone lesions (13, 14, 15, 16, 17). Because of the rarity of ALCL presented as bone lesions, it remains to be determined whether ALCL involving primarily bones has distinctive biologic and clinical features.
In this study, we described the clinicopathologic features of six cases of ALCL primarily presented as one or more bone lesions. All patients were found to have only bone involvement in the initial clinical assessment. We also compare these cases with those previously published in the literature to determine if these neoplasms have biological and clinical features different from ALCL of nodal or other nonbone extranodal sites.
MATERIALS AND METHODS
Cases
The clinical and pathologic data of six cases of ALCL that presented as bone lesions were compiled and analyzed. The disease of all patients was staged with chest, abdominal, and pelvic computed tomography scans. Bone marrow biopsy was performed and the International Prognostic Index (IPI) was also available for all cases. Lymphomas were classified in accordance with the REAL and newly proposed WHO classifications using immunohistochemical and morphologic features (9, 10).
Immunohistochemistry
Immunohistochemical studies were performed using routinely fixed/processed archival tissues. Monoclonal antibodies specific for the following antigens were used: CD1 (O10), prediluted (Immunotech, Marseille, France); CD3, 1:50; CD4, 1:50; CD8, 1:50 (Novocastra Laboratories, Newcastle-on-Tyne, UK); CD2 (MT910), 1:50; CD20 (L26), 1:100; CD30 (BerH2), 1:5; EMA (E29), 1:20; ALK-1, 1:25 (DAKO, Kyoto, Japan); Granzyme B, 1:30 (Monosan, Uden, the Netherlands); TIA-1, 1:50 (Coulter Immunology, Hialeah, FL). Tissue sections were mounted on precleaned slides (Superfrost, Matsunami Glass Industries, Ltd., Tokyo, Japan), baked at 56° C for 60 min, deparaffinized with xylene, and rehydrated with ethanol to distilled water. A prestaining heat-induced antigen retrieval method was used, as previously described (18). Briefly, tissue sections were placed in sodium citrate buffer (0.01 mol/L, pH 6.0) and heated in a microwave oven for 10 min. An automated immunostainer (Ventana Biotek Medical Systems, Tuczon, AZ) was used. Reactivity was detected by an avidin-biotin immunoperoxidase method.
In Situ EBV Hybridization
In situ hybridization studies to detect Epstein-Barr virus RNA were performed using a 30-base oligonucleotide complementary to a portion of EBER1 genome that is actively transcribed (up to 107 copies per cell) in latently infected cells. The methods have been detailed elsewhere (19).
RESULTS
Case Reports
Patient 1
This 4-year-old Japanese boy presented with fever and painful swelling of the right wrist. His past medical history was unremarkable. Initial physical examination was essentially noncontributory, other than the swelling of the right wrist. Radiologic examination revealed two osteolytic lesions, one 2-cm lesion located in the epiphysis of the right radius, and one 3-cm lesion located in the diaphysis of the right tibia. Laboratory studies showed mild leukocytosis and an elevated erythrocyte sedimentation rate. On the basis of these findings, the clinical differential diagnosis included eosinophilic granuloma and osteomyelitis. This patient underwent biopsy of the tibial lesion, which revealed anaplastic large cell lymphoma. Staging studies revealed mild bilateral cervical lymphadenopathy. Bone marrow examination was negative. The IPI was 2. After a course of combination chemotherapy followed by autologous bone marrow transplant, the disease relapsed with generalized lymphadenopathy, hepatosplenomegaly, and bone marrow involvement. He died two years after the initial diagnosis.
Patient 2
This 24-year-old Japanese female presented with fever and right buttock pain. Her past medical history was unremarkable. Physical examination revealed no evidence of lymphadenopathy or hepatosplenomegaly. Radiologic studies revealed a solitary 7-cm osteolytic lesion of the left iliac bone. Laboratory studies revealed an elevated erythrocyte sedimentation rate and leukocytosis. The clinical differential diagnosis included osteomyelitis and giant cell tumor. A biopsy was done, which revealed anaplastic large cell lymphoma. Staging studies did not reveal evidence of disease elsewhere. Bone marrow examination was negative. The IPI was 1. Despite a course of combination chemotherapy (cyclophosphamide, daunorubicin, vincristine, prednisone; CHOP), she died of disease one year after the initial presentation.
Patient 3
This 27-year-old Caucasian woman presented with a 3-month history of right hip pain. She was otherwise healthy and her medical history was noncontributory. Radiologic studies revealed a 3-cm destructive lesion in the right iliac wing. The surrounding cortical bone was focally broken through by this lesion, with periosteal reaction and edema. The clinical differential diagnosis included Ewing’s sarcoma and other small round cell tumors. A biopsy was performed, which revealed anaplastic large cell lymphoma. Bone marrow biopsy showed no evidence of lymphoma. Further staging studies showed no evidence of disease elsewhere. The IPI was 1. This patient received combination chemotherapy (six courses of CHOP) followed by radiotherapy, and she was in remission 8 mo after the initial presentation.
Patient 4
This 30-year-old Japanese man presented with tenderness of the right sixth costal bone. His past medical history was unremarkable. Physical examination was negative. Radiologic studies revealed two osteolytic lesions, a 1-cm lesion located in the right sixth rib, and one 4-cm lesion located in the right iliac bone. Laboratory studies were unremarkable. The clinical impression was that of a primary bone tumor. A biopsy of the rib lesion was performed, which revealed anaplastic large cell lymphoma. Staging procedure did not reveal evidence of disease elsewhere. Bone marrow was negative for neoplasm. The IPI was 1. After a course of combination chemotherapy (CHOP) and autologous peripheral blood stem cell transplantation, the patient was alive with disease in the rib and iliac bone 2 years after the initial presentation.
Patient 5
This 50-year-old Japanese man presented with fever and back pain. His past medical history was unremarkable. Physical examination was also unremarkable; no lymphadenopathy or hepatosplenomegaly was identified. Radiologic studies revealed multiple osteolytic lesions present in multiple levels of the vertebral bones (T6, L2, L3, and S1). Abnormal laboratory findings included elevated lactate dehydrogenase and C-reactive protein. The clinical diagnosis was metastatic tumor to bone of unknown origin. A biopsy of the sixth thoracic vertebra was performed and the pathological diagnosis was anaplastic large cell lymphoma. Staging procedure did not reveal evidence of disease elsewhere. Bone marrow was negative for lymphoma. The IPI was 3. He underwent a course of combination chemotherapy (CHOP) and irradiation, and he was alive with disease involving lumbar (L2, L3) and sacral (S1) vertebrae two years after the initial presentation.
Patient 6
This 63-year-old Japanese man presented with lower back pain. His past medical history was unremarkable. Physical examination was negative; no lymphadenopathy or hepatosplenomegaly was identified. Radiologic investigations revealed a 5-cm osteolytic lesion involving the first lumbar vertebrae (Fig. 1). Abnormal laboratory findings included elevated C-reactive protein. The clinical diagnosis was metastatic tumor of unknown origin. A biopsy of the L1 lesion was performed, which revealed anaplastic large cell lymphoma. Bone marrow was negative for lymphoma. Further staging procedures did not reveal disease elsewhere. The IPI was 2. He did not respond to one course of combination chemotherapy CHOP), and he died of disease 3 mo after diagnosis.

Magnetic resonance imaging (MRI) of the osteolytic bone lesion in the lumbar vertebra (L5) in Case 6.
The clinical data of these six cases of ALCL primarily presented as one or more bone lesion are summarized in Table 1 and as follows. (1) The mean patient age in this series was 33 years (range, 4 to 63 years). (2) Axial bones were involved in most (5/6, 83%) cases. (3) The mean of the International Prognostic Index (IPI) for these patients was relatively low (mean, 1.67; range, 1 to 3). (4) Despite treatment, the overall prognosis was relatively poor: three of six died of disease within 2 years of diagnosis and two of six were alive with evidence of disease (follow-up, 6 mo to 2 years).
Histologic Findings
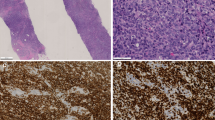
The bone lesions of all cases were composed of diffuse sheets of lymphoma cells (Fig. 2A). Reactive bone was found at the periphery of the tumors. In Patient 3, the tumor penetrated the bone and infiltrated the surrounding skeletal muscle, with edema of the surrounding soft tissues. On higher magnification, the lymphoma cells were pleomorphic (Fig. 2B). Cells with a central eosinophilic body (so-called hallmark cells) (20) were present in all cases. Reed-Sternberg-like cells were frequently found in Patient 4. The mitotic rate was generally high (more than 15 per ten 400 × high-power fields); focal necrosis and admixed neutrophils were commonly seen.

A, a low-magnification view of the bone lesion, showing diffuse infiltration by a population of mononuclear tumor cells (Case 4; hematoxylin and eosin stain, original magnification, 40 ×). B, a high-magnification view of the bone lesion. The tumor cells were large and pleomorphic, with frequent mitoses (Case 4; hematoxylin and eosin stain, original magnification, 200 ×).
Immunohistochemical and In Situ Hybridization Findings
The immunohistochemical results are summarized in Table 2 and partly illustrated in Figure 3. CD30 expression was identified in most (more than 90%) tumor cells in all cases, and the staining was localized to the perinuclear regions as well as the cell membrane. ALK-1 staining was observed in Patients 1, 3, and 6. The staining was largely cytoplasmic, although a weaker nuclear staining was also observed in all three cases. EMA and granzyme B were expressed in most tumor cells (more than 50% tumor cells) in all six cases. One case was of T-cell lineage, positive for CD3, and one case expressed the T-cell-associated antigen CD4. The remaining four cases were considered null-cell type. All cases were negative for CD20 and CD8, and only one of five tested cases was positive for TIA.

Immunohistochemical characterization of the tumor (Case 6) revealed strong reactivity in the majority of tumor cells with CD30 (A), epithelial membrane antigen (B), granzyme B (C), and ALK-1 (D) (immunoperoxidase stain, original magnification, 400 ×).
In situ hybridization for Epstein Barr virus was performed; all five cases tested were negative.
DISCUSSION
ALCLs presented as bone lesions are uncommon; relatively few such cases have been briefly mentioned in several clinicopathologic studies of ALCL (21, 22, 23). We identified 10 cases with more detailed pathologic and immunologic characterization in the literature, and they are summarized in Table 3. The mean patient age of these patients is 33 years (range, 8 to 55 years). One patient (Case 10) was HIV-positive. In this report, we add six additional cases of ALCL primarily presented as bone lesions.
At initial presentation, all six cases in this series had evidence of disease confined to the bone. Staging studies showed that all except one patient had no evidence of disease elsewhere. The only patient (Patient 1) with extra-skeletal disease was found to have mild cervical lymphadenopathy, which is more consistent with metastasis than primary nodal disease. Similar to the previously published 10 cases, the most common initial clinical diagnosis for these six cases included eosinophilic granuloma and osteomyelitis in the younger patients and metastatic malignancies in the older patients. Radiographically, all tumors presented as osteolytic bone lesions, with three of six showing involvement in multiple skeletal sites. They were typically composed of diffuse sheets of large pleomorphic lymphoma cells. Tumor cells with a central eosinophilic body (the so-called hallmark cells) (20) were identified in all cases. These tumors often expressed granzyme B (6/6, 100%), EMA (6/6, 100%), and ALK-1 (3/6, 50%), but showed no evidence of Epstein-Barr virus.
When the features of the current cases and those published previously are compared (Table 3), certain common features emerge. First, most of the patients of primary bone ALCL are young to middle-aged: the mean age is 21.4 (range, 8 to 55) in the 10 published cases, and the mean patient age in our study is 33.0 years (range, 4 to 63 years). Second, these tumors appear to have a predilection for the axial bones: five of six current cases and 8 of 10 cases of the previously reported cases involve the pelvic bone and/or vertebral column. Third, the overall prognosis of these neoplasms is poor. In this series, despite the relatively low IPI (mean, 1.67), five of six patients did not have a favorable outcome: three of six died of disease within 2 years after the initial presentation and two of six lived with evidence of disease during a follow-up period of 6 mo to 2 years. As shown in Table 3, most of those previously reported cases also had a relatively poor clinical outcome. All four cases described by Jones et al. (15) were patients with stage 1E or II diseases; three died of disease within 23 mo of diagnosis. Lastly, despite the fact that the expression of the ALK-1 protein is generally associated with young patient age and better prognosis in conventional ALCL (5), two of three cases with ALK-1 positivity in this series (Case 1 and 6) died of disease within 2 years after the initial presentation. The patients of all three cases described previously showing ALK-1/p80NPM/ALK reactivity (Table 3, Case 1, 5, and 6) also died of disease within 2 years of diagnosis. Thus, the view that ALK-1/p80NPM/ALK is a favorable prognostic predictor, as shown in ALCL of nodal and nonbone extranodal sites, does not appear to be valid for primary bone ALCL.
Another interesting finding was the frequent expression of cytotoxic proteins, with six cases expressing granzyme B. In a few recent studies, a subset of ALCL was found to be positive for these molecules, although the frequency of expression varies among studies (24, 25, 26, 27). Based on these findings, it has been proposed that most cases of ALCL originate from lymphocytes with cytotoxic potential. However, the correlation between cytotoxic protein expression and ALK-1/p80NPM/ALK is controversial. For instance, the expression of cytotoxic proteins was found to be correlated with p80NPM/ALK in one study (26), but not in another study (24). Importantly, the expression of cytotoxic proteins does not appear to have any significant impact on prognosis (27). The biological and clinical significance of granzyme B in the ALCL of this study is unclear.
In summary, we characterized six unusual cases of ALCL primarily presented as osteolytic bone lesions, with a predilection for the axial bones. Most (4/6) tumors were of null-cell lineage; all cases tested were positive for granzyme B, and five of six have had relatively poor clinical outcome. Our findings also suggest that, unlike nodal ALCL, ALK-1 positivity is not a favorable prognostic feature for patients with primary bone ALCL. These findings further support the concept that the clinical behavior and pathogenesis of ALCL may vary according to the involved anatomic sites.
References
Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G, et al. The expression of Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence of Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985; 66: 848–858.
Kadin ME . Primary Ki-1-positive anaplastic large-cell lymphoma: a distinct clinicopathologic entity. Ann Oncol 1994; 5: S25–S30.
Kadin ME, Morris SW . The t(2;5) in human lymphomas. Leuk Lymphoma 1998; 29: 249–256.
Nakamura S, Shiota M, Nakagawa A, Yatabe Y, Kojima M, Motoort T, et al. Anaplastic large cell lymphoma: a distinct molecular pathologic entity. A reappraisal with special reference to p80NPM/ALK expression. Am J Surg Pathol 1997; 21: 1420–1432.
Shiota M, Nakamura S, Ichinohasama R, Abe M, Akagi AT, Takeshita M, et al. Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK. A distinct clinicopathologic entity. Blood 1995; 86: 1954–1960.
Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleoplasmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997; 89: 1394–1404.
Noorduyn LA, Bruin PC, van Heerde P, van de Sandt MM, Ossenkoppele GJ, Meijer CJ . Relation of CD30 expression to survival and morphology in large cell B cell lymphoma. J Clin Pathol 1994; 47: 33–37.
Warnke RA, Weiss LM, Chan JKC, Cleary ML, Dorfman RF . Malignant lymphoma, diffuse, large cell and variants. In: Rosai J, editor. Atlas of tumor pathology. Tumors of the lymph nodes and spleen. Washington, DC: Armed Forces Institute of Pathology; 1994. p.153–220.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361–1392.
Jaffe ES, Harris NL, Diebold J, Muller-Hermelink HK . World Health Organization Classification of lymphomas: a work in progress. Ann Oncol 1998; 9 (Suppl 5): S25–S30.
DeCoteau JF, Butmarc JR, Kinney MC, Kadin ME . The t(2;5) chromosomal translocation is not a common feature of primary cutaneous CD30+ lymphoproliferative disorders: comparison with anaplastic large cell lymphoma of nodal origin. Blood 1996; 87: 3437–3441.
Ostrowski ML, Unni KK, Banks PM, Shives TC, Evans RG, O’Connell MJ, et al. Malignant lymphoma of bone. Cancer 1986; 58: 2646–2655.
Suzukawa K, Kojima H, Mori N, Mukai HY, Hori M, Komeno T, et al. Anaplastic large cell lymphoma of null-cell type with multiple bone involvement. Ann Hematol 1998; 77: 287–290.
Ishizawa K, Okabe H, Matsumoto K, Hukuda S, Hodohara K, Ota S . Anaplastic large cell Ki-1 lymphoma with bone involvement: report of two cases. Virchows Arch 1995; 427: 105–110.
Jones D, Kraus MD, Dorfman DM . Lymphoma presenting as a solitary bone lesion. Am J Clin Pathol 1999; 111: 171–178.
Chan JKC, Ng CS, Hui PK, Leung WT, Sin VC, Lam TK, et al. Anaplastic large cell Ki-1 lymphoma of bone. Cancer 1991; 68: 2186–2191.
Piira TA, Ries K, Kjeldsberg CR, Perkins SL . Anaplastic large cell lymphoma presenting primarily in bone in a patient with AIDS. Hematol Pathol 1994; 8: 111–116.
Battifora H, Alsabeh R, Jenkins KA, Gown A . Epitope retrieval (unmasking) in immunohistochemistry. Adv Pathol Lab Med 1995; 8: 101–118.
Ambinder RF, Browning PJ, Lorenzana I, Leventhal BG, Cosenza H, Mann RB, et al. Epstein-Barr virus and childhood Hodgkin’s disease in Honduras and the United States. Blood 1993; 81: 462–467.
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe M, et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 1998; 91: 2076–2084.
Penny RJ, Blaustein JC, Longine JA, Pinkus GS . Ki-1 positive large cell lymphoma, a heterogeneous group of neoplasms. Morphologic immunophenotypic, genotypic and clinical features of 24 cases. Cancer 1991; 68: 362–373.
Greer JP, Kinney MC, Collins RD, Salhany KE, Wolff SN, Hainsworth JD, et al. Clinical features of 31 patients with Ki-1 anaplastic large cell lymphoma. J Clin Oncol 1991; 9: 539–547.
Chott A, Kaserer K, Augustin I, Veseley M, Heinz R, Oehlinger W, et al. Ki-1-positive large cell lymphoma. A clinicopathologic study of 41 cases. Am J Surg Pathol 1990; 14: 439–448.
Krenacs L, Wellmann A, Sorbara L, Himmelmann AW, Bagdi E, Jaffe ES, et al. Cytotoxic cell antigen expression in anaplastic large cell lymphomas of T- and null-cell type and Hodgkin’s disease: evidence for distinct cellular origin. Blood 1997; 980–989.
Foss HD, Demel G, Anagnostopoulos I, Araujo I, Hummel M, Stein H . Uniform expression of cytotoxic molecules in anaplastic large cell lymphoma of null/T cell phenotype and in cell lines derived from anaplastic large cell lymphoma. Pathobiology 1997; 65: 83–90.
Felgar RE, Salhany KE, Macon WR, Pietra GG, Kinney MC . The expression of TIA-1+ cytolytic-type granules and other cytolytic lymphocyte-associated markers in CD30+ anaplastic large cell lymphoma (ALCL): correlation with morphology, immunophenotype, ultrastructure, and clinical features. Hum Pathol 1999; 30: 228–236.
Dukers DF, ten Berge RL, Oudejans JJ, Pulford K, Hayes D, Misere JF, et al. A cytotoxic phenotype does not predict clinical outcome in anaplastic large cell lymphomas. J Clin Pathol 1999; 52: 129–136.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nagasaka, T., Nakamura, S., Medeiros, L. et al. Anaplastic Large Cell Lymphomas Presented as Bone Lesions: A Clinicopathologic Study of Six Cases and Review of the Literature. Mod Pathol 13, 1143–1149 (2000). https://doi.org/10.1038/modpathol.3880211
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.3880211
Keywords
This article is cited by
-
Freiburg Neuropathology Case Conference
Clinical Neuroradiology (2020)
-
Primary ALK-negative anaplastic large cell lymphoma with extensive bone involvement mimicking multiple myeloma and metastatic carcinoma
International Journal of Hematology (2019)
-
An Immunophenotypic and Molecular Study of Primary Large B-Cell Lymphoma of Bone
Modern Pathology (2001)
