
Abstract
Biomphalaria glabrata is the main intermediate host of Schistosoma mansoni in America and one of the most intensely studied species of freshwater snail, yet very little is known about its population biology. Here, we used seven highly polymorphic microsatellite loci to analyse genetic diversity in populations from three regions (Lesser Antilles, Venezuela and southern Brazil). Considerable genetic variation was detected, with an average (s.d.) H0 = 0.32 (0.24). More diversity per population was found in the Valencia lake basin in Central Venezuela, which suggests an influence of dispersal (via inter-population connectivity) on the restoring of genetic diversity after the demographic bottlenecks recurrently experienced by populations. A marked population structure was detected and there seems to be a relationship between mean differentiation and genetic diversity within regions. There is also a significant isolation-by-distance pattern. The Lesser Antilles populations appear clearly differentiated from the rest, which suggests a single colonisation event followed by local radiation within these islands or multiple colonisation events from the same source area. Our results indicate that B. glabrata essentially cross-fertilises, with little variation in selfing rates among populations. However, significant deficits in heterozygotes and linkage disequilibria were detected in two Venezuelan populations suggesting a mixture of at least two different genetic entities, probably with differences in their respective mating systems.
Similar content being viewed by others

Introduction
A major aim of population genetics studies is to explain the distribution of genetic diversity within and among populations. This distribution may reflect a given mutation-selection-drift regime at equilibrium. However, recently, short-scale demographic variations and dispersal events (eg, extinction and re-colonisation) have attracted increasing interest because they influence both within-population diversity and differentiation among populations (Wade and McCauley, 1988; McCauley, 1991; Rousset, 2001). Empirical evidence for these effects has been found in forked fungus beetles (Whitlock, 1992) and in Silene dioica (Giles and Goudet, 1997). The mating system, especially self-fertilisation, also correlates with diversity within and among populations (see Charlesworth et al, 1993 for theoretical considerations; Jarne, 1995 and Hamrick and Godt, 1989 for empirical examples on animal and plant species, respectively).
Surprisingly few studies have attempted to depict the joint influence of short-term demographic instability and mating system in natural populations, probably due to a lack of adequate model organisms. In this respect, freshwater snails, especially tropical pulmonate species, can be excellent models (Städler and Jarne, 1997). (i) This group of organisms commonly live in well-defined habitats fragmented in space, such as rivers and lakes (Jarne and Delay, 1991), with low connectivity among sites, which should limit dispersal. Water availability varies in time because of seasonal climatic cycles, which can lead to large fluctuations in population size and even to local extinction (Sturrock, 1973; Pointier et al, 1993; Brown, 1994). Re-colonisation of empty habitats is generally fast, and may sometimes be facilitated by human activities. More generally, freshwater snails are extremely efficient colonisers at a large geographical scale (Madsen and Frandsen, 1989; Pointier, 1999). Introduction into new habitats, such as islands, might be associated with a reduction in genetic diversity as a consequence of genetic bottlenecks. (ii) Freshwater pulmonate snails are simultaneous hermaphrodites, and reproduction by both self- and cross-fertilisation is possible (Jarne et al, 1993). However, selfing rates seem to have a bimodal distribution in this group, with most species being preferentially selfers or outcrossers.
We focus here on Biomphalaria glabrata, a South-American pulmonate freshwater snail. This species is interesting not only as a model system (see above) but also because it is the major vector of Schistosoma mansoni, the causative agent of Schistosomiasis in the New World. The few available studies of genetic variation in natural populations were based on either allozymes (eg, Mulvey et al, 1988) or RAPD (eg, Sire et al, 1999) markers, and therefore suffer from either low polymorphism or technical limitations. They also included few populations sampled at limited geographic scale. Little is therefore known on the demography and mating system of B. glabrata, and it is even possible that this taxon encompasses several genetically heterogeneous entities (Woodruff and Mulvey, 1997; Mavárez et al, 2002b). Microsatellite markers have recently been described in this species (Jones et al, 1999; Mavárez et al, 2000) and proved to be useful for small-scale genetic analyses of population structure and mating system in central Venezuela (Mavárez et al, 2002a). We here use these markers to describe the genetic diversity of B. glabrata at the scale of its distribution area, from Venezuela and the Lesser Antilles (north) to Brazil (south). Note that a similar analysis has been performed with nuclear and mitochondrial DNA sequences in a companion paper (Mavárez et al, 2002b). These data are used to answer three questions: (i) What does genetic diversity within and among populations tell us about the past and present demography and dispersal of snail populations? (ii) What is the mating system of B. glabrata? (iii) Can B. glabrata be considered as a single homogeneous genetic entity across its distribution area?
Materials and methods
Species studied
Biomphalaria glabrata is a tropical species, widely distributed from south-eastern Brazil to the northern part of Haiti (Paraense, 2001), where it is commonly found inhabiting various freshwater habitats (eg, ponds and ditches; Pointier, 1976; Mulvey et al, 1988). These habitats have a patchy distribution and dispersal of snails between localities can be very limited (Mavárez et al, 2002a). Demographic instability, including large fluctuations of population size and frequent local extinction/re-colonisation phenomena, has been described in this species (Sturrock, 1973; Pointier and Theron, 1979). This results in marked genetic differentiation (Mulvey and Vrijenhoek, 1982; Mulvey et al, 1988), even at local scale (Mavárez et al, 2002a). B. glabrata is a simultaneous hermaphrodite which can reproduce by both self and cross-fertilisation (Vianey-Liaud, 1997). The few genetic studies conducted showed that selfing rates are low (see Vianey-Liaud, 1997; Mavárez et al, 2002a). However these studies included few populations or laboratory strains.
Snail sampling
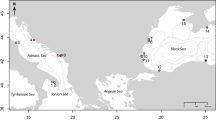
A total of 436 adult individuals were collected in one locality from Brazil, 14 from Venezuela and three from the Guadeloupe archipelago in the Lesser Antilles (Table 1 and Figure 1). Five populations (VE2, VE3, VE5, VE8, VE10) from central Venezuela have been analysed previously (Mavárez et al, 2002a). These localities represent very different habitats types (ie, small rivers, canals, lakeshores and ponds) experiencing different seasonal regimes. They can be classified as permanent or temporary with regard to temporal water availability (Table 1). Snails were collected using a scoop or a lending net. They were then killed in 80°C water for 30 sec, and preserved in 96% ethanol before genetic analysis.

Distribution of 18 populations of Biomphalaria glabrata sampled from Brazil, Venezuela and the Lesser Antilles. The distribution area of B. glabrata is indicated in grey on the left bottom map.
DNA extraction and microsatellite typing
DNA was extracted from an individual’s foot using the QIAmp tissue kit (Qiagen). The variability was analysed at seven polymorphic microsatellite loci (Table 2) which were cloned in B. glabrata, and showed non-ambiguous amplification patterns. Detailed information about loci, PCR amplification and scoring can be found in Mavárez et al (2000) for loci BgC7, BgC8, BgE2, BgE4 and BgE5, and in Jones et al (1999) for loci BgU1 and BgU2. Briefly, individual microsatellite genotypes were obtained using either of two methods. In the first one, PCR amplification included γ-33P-labelled primers, and was followed by electrophoresis in acrylamide gels (Mavárez et al, 2000). The second method included PCR-amplification with fluorescent-labelled primers, and electrophoresis on a ABI 310 automatic genetic analyser using the manufacturer’s protocols. Some individuals were analysed using both methods in order to correct for potential differences in allele scoring.
Diversity within populations
Standard parameters, including the number of polymorphic loci, allelic richness (Petit et al, 1998), observed heterozygosity (Ho) and gene diversity (He corrected for small sample size; Nei, 1987) were used to describe the genetic polymorphism for each population/locus combination. Differences in mean allelic richness and mean He between geographic areas (ie, the Lesser Antilles, western Venezuela and central Venezuela) were tested using Mann-Whitney U tests (Sokal and Rohlf, 1995). Deviations from the genotypic proportions expected at Hardy-Weinberg equilibrium at each locus and from genotypic equilibrium between pairs of loci were tested in each population using exact tests (Rousset and Raymond, 1995). Departures from Hardy-Weinberg equilibrium over all loci were evaluated using Fisher’s method for combining probabilities (Sokal and Rohlf, 1995). All calculations and tests were performed using the GENEPOP version 3.1d package (Raymond and Rousset, 1995b), with the exception of allelic richness that was calculated using FSTAT 2.9.3.1 package (Goudet, 2001). The unbiased estimator of Wright’s inbreeding coefficient FIS, f, was calculated following Weir and Cockerham (1984). Its significance was assessed using a permutation method (10000 permutations) implemented in the GENETIX 4.01 package (Belkhir et al, 2000). The selfing rate s was estimated according to the formula ŝ = 2f/(1 + f). This expression is independent of the mutation model of the markers used (Rousset, 1996), and has been shown to be only slightly biased when compared to more accurate methods such as those based on progeny-arrays (Viard et al, 1997a).
Differentiation among populations
Two approaches were followed for analysing the distribution of variability among populations. (i) The per-locus differentiation for both all population pairs and all populations was tested using exact tests (Raymond and Rousset, 1995a; Goudet et al, 1996), and Wright’s FST was estimated using the estimator θ (Weir and Cockerham, 1984). Isolation-by-distance was tested using a Mantel-like procedure contrasting multilocus θ-values and geographic distances between population pairs (Rousset, 1997). Under this model, the slope of the linear regression between θ/(1−θ) and the logarithm of distance (in a two-dimensional habitat) for each population pair is expected to be equal to 1/4πDσ2, where D and σ2 represent the population density and variance in dispersal distance of individuals respectively (Rousset, 1997). The formula DAB = R . Arccos {sin(π/2 − LA) . sin(π/2 − LB) . cos(lA − lB) + cos(π/2 − LA) . cos(π/2 − LB)}, where R is the radius of the earth, LA and LB the latitudes and lA and lB the longitudes of populations A and B in radians, was used to calculate the geographic distances between populations. Geographic coordinates for Venezuelan and insular populations were obtained with a global positioning system. Coordinates for the Brazilian population were estimated from a map. The calculations were performed using the GENEPOP 3.1d package (Raymond and Rousset, 1995b). (ii) Genetic affinities among populations were described using a factorial correspondence analysis (FCA) on multilocus genotypes as described in the GENETIX 4.01 package (Belkhir et al, 2000). A hierarchical analysis of molecular variance (AMOVA; Excoffier et al, 1992) was also conducted on allele frequencies, with populations ranked within regions (Lesser Antilles, western Venezuela and central Venezuela). The AMOVA analyses were performed using ARLEQUIN 1.1 (Schneider et al, 1999).
Results
Information about the polymorphism at the seven loci used averaged over the 18 populations studied is given in Table 2. A significant correlation was found between the longest uninterrupted string of repeated motifs in the core sequence of the cloned allele and the mean number of alleles across loci (Spearman r = 0.78, P = 0.01). Loci BgE2, BgU2 and BgE4 failed to amplify in populations VE12 and VE14, the Lesser Antilles populations and the Brazilian population, respectively. Estimates of genetic variation were highly variable among regions (Table 3). For example, populations from central Venezuela showed the highest genetic diversity, although they include two poorly variable populations (VE2 and VE4). On the other hand, the Brazilian population was monomorphic at the five amplified loci. However, no significant difference in genetic diversity estimates among regions were detected (Mann-Whitney’s U test on allelic richness and He, P > 0.302 in both situations), perhaps as a consequence of limited sample sizes.
Hardy-Weinberg disequilibrium (heterozygote deficiency) was found in 18, out of 78, locus-population combinations. However, loci BgU2 and BgE2 accounted for nine of these deviations. A likely explanation is the occurrence of null alleles, since no amplification were obtained at both loci for some individuals (see above). When these loci were discarded, most deficits in heterozygotes (eight) were detected in the VE6 and VE13 populations, respectively. As expected, significant departures from Hardy-Weinberg equilibrium (multilocus test, P < 0.0001) were detected in these two populations only. The associated selfing rates were 0.69 and 0.80 (Table 3). The mean f value (s.d.) for the other populations was −0.02 (0.10), and 0.05 (0.12) when including BgE2 and BgU2. Accordingly, the mean selfing rate was very low (ŝ = 0.10; see Table 3). Genotypic disequilibria were tested in 172 locus pairs – population combinations, 29 of which were significant at the 0.05 level, which is three times higher than what is expected by chance alone. Populations VE6 and VE13 accounted for 52% and 41% of these combinations, respectively. After sequential Bonferroni correction, 15 tests remained significant, all in VE13 (6) and VE6 (9) populations.
These two populations deserve closer examination. VE13 is made of two groups. Group 1 contains five individuals for which BgE2 could not be amplified, and sharing the same homozygous genotype at the other six loci. Group 2 includes the remaining 19 individuals, each exhibiting unique multilocus genotypes. No heterozygote deficiency was detected in group 2. Only in one individual and one locus of group 2 could we find an allele shared with the group 1 multilocus genotype. The situation in VE6 is very similar, with 19 individuals sharing exactly the same multilocus genotype as in group 1 of population VE13, although the populations are separated by 250 km. The other five individuals in VE6 each exhibited unique multilocus genotypes, and again, a single allele was shared by a single individual with the other individuals. Hardy-Weinberg tests were not performed in this group because of low sampling size. Finally, we note that the alleles found in the common multilocus genotype found in both VE6 and VE13 are on the whole very rare in other populations. They reach a maximum frequency of 0.08 in western Venezuelan populations; and they simply do not exist elsewhere.
Population differentiation, tested across all populations both for each locus and over all loci, was highly significant (exact tests, P < 0.0001 in all situations). Moreover, 98.6% of single-locus tests between population pairs were statistically significant at the 0.05 level. Only the less polymorphic loci (BgC7 and Bgc8) showed some non-significant tests, which is consistent with the expected relationship between genetic polymorphism and the power of differentiation tests (Goudet et al, 1996). The multilocus θ values between pairs of populations ranged between 0.07 and 1.00 with an overall mean (s.d.) value of 0.59 (0.21). The corresponding values within regions were 0.62 (0.17), 0.53 (0.11) and 0.44 (0.22) for the Lesser Antilles, western Venezuela and central Venezuela respectively. The Mantel procedure test between genetic differentiation and geographical distance for pairs of populations was highly significant (P < 10−5), indicating isolation-by-distance (see Figure 2). The Dσ2 product estimated from the regression slope was 0.0012 individuals.

Isolation-by-distance  as a function of the logarithm of geographic distance) in 18 populations of Biomphalaria glabrata from Brazil, Venezuela and the Lesser Antilles.
as a function of the logarithm of geographic distance) in 18 populations of Biomphalaria glabrata from Brazil, Venezuela and the Lesser Antilles.
Figure 3 shows the two-dimensional diagram derived from the FCA analysis on multilocus genotypes. The diagram shows that Lesser Antilles populations appear clearly differentiated from all the other populations (mostly on the basis of axis 1). The eastern Venezuela population also seems to be differentiated from the rest. All the other populations cluster in the same section of the diagram. Note that VE6 and VE13 populations cluster here with other populations from western and central Venezuela. However, the group sharing the same multilocus genotype is clearly differentiated from the rest (data not shown). The results of the hierarchical analysis of variance indicate that the ‘within populations’, ‘among populations within regions’ and ‘among regions’ sources of variation explain 40%, 38% and 22% of the variance in allelic frequencies, respectively (P < 10−4 for the three groups).

A two-dimensional diagram representing the relationships between 17 populations of Biomphalaria glabrata from the Lesser Antilles and Venezuela (the Brazilian populations is not included, see text), based on a factorial correspondence analysis on multilocus genotypes. Only the first two axes are represented (with the percent of variance explained within parentheses).
Discussion
Genetic differentiation among populations and isolation-by-distance
Our results indicate that the B. glabrata populations studied here cannot be considered as being drawn from the same gametic pool. Even the geographically close populations from Lake Valencia (eg, VE3, VE5 and VE8) appear genetically differentiated (mean θ = 0.083). This suggests that dispersal is limited in B. glabrata, in agreement with previous studies in this species (Mulvey and Vrijenhoek, 1982; Mulvey et al, 1988; Mavárez et al, 2002a), as it is in general in many freshwater snails (Viard et al, 1997b; Charbonnel, 2001; Meunier et al, 2001) and other freshwater invertebrates (Freeland et al, 2000; Pálsson, 2000). Whether low dispersal alone may explain the observed genetic differentiation deserves further consideration. The populations studied indeed experience strong variation in size, as well as extinction/re-colonisation (see below). Theoretical models have shown that these processes enhance population differentiation under a wide range of parameter values (McCauley, 1991; Harrison and Hastings, 1996). Their influence has been documented in natural populations of freshwater invertebrates (Charbonnel et al, 2002) and plants (Giles and Goudet, 1997), and could well work here.
When gene flow is spatially limited, an equilibrium may develop between gene flow and genetic drift, resulting in isolation-by-distance (Rousset, 1997). It is therefore interesting that such a pattern was detected in our study, despite the fact that the water bodies are likely to be discontinuously connected by gene flow. This pattern is consistent with the AMOVA results, showing a significant pattern of hierarchical differentiation. The Dσ2 estimate inferred from Rousset (1997) method (0.0012 individuals) appears very low when compared with our previous estimations for Lake Valencia (1.66 individuals) and its basin (0.56 individuals) (Mavárez et al, 2002a). This is not unexpected given the geographic scale and the isolation of most populations. Indeed the dispersion mechanisms at the scale of the distribution area may be very different from dispersal within a lake or a basin. However, the difference between estimates should be interpreted with caution, for example because the scale at which the method was used here is likely to be too large for mutation to be neglected (Rousset, 1997, 2001).
That isolation-by-distance was detected is globally consistent with the AMOVA and FCA analyses. An interesting result is the association of the Lesser Antilles populations and their differentiation vis-à-vis all the other population analysed, which suggests a single colonisation event followed by local radiation within these islands or repeated introductions from a single source. However, we note that except for the Lesser Antilles and eastern Venezuela populations, most of the populations analysed appear clustered in Figure 3. This may result from the low percent of variance explained by the first axes of the FCA. We have conducted a more powerful analysis of intra-specific variation using phylogenetically informative markers such as mitochondrial 16S and nuclear ITS-2 gene sequence variation among populations of B. glabrata from a large part of its distribution area (Mavárez et al, 2002b).
Genetic variation within populations
Our studies reveal that B. glabrata is a highly variable species, ranking as the most variable (when using microsatellites) freshwater pulmonate species studied so far, at least in the northern part of its distribution area (Table 4). Caution is required here since few studies have been performed. The geographic location of samples seems to play a role here. Populations from central Venezuela were more variable than the other populations from Venezuela, the Lesser Antilles and Brazil. There are several non-exclusive explanations to these differences: (i) Historical factors. Except for populations from central Venezuela (Valencia lake basin), most populations analysed here occupy either recently created habitats, or habitats associated with human activities, such as canals and artificial ponds. Populations were presumably founded by a low number of colonisers. Moreover, populations from the Lesser Antilles were probably founded very recently, at least no earlier than the first human occupations, a few thousand years ago. The habitats before human occupation (dense forest) were probably not favourable to freshwater pulmonate snail species. It remains possible that the colonisation of new habitats, especially in the Lesser Antilles, was associated with bottlenecks. (ii) Current forces. Previous work on freshwater snails has highlighted the role of local gene flow on diversity (Viard et al, 1997b; Charbonnel et al, 2002; Mavárez et al, 2002a). In other words, populations from central Venezuela might be more interconnected than elsewhere, allowing for the maintenance of more diversity (Barton and Whitlock, 1997). Populations outside central Venezuela are indeed patchily distributed, with limited opportunity for immigration, while populations from central Venezuela are connected to the large population of Lake Valencia (Mavárez et al, 2002a). For example, the VE4 and GU1 populations are the only localities inhabited by B. glabrata within their respective geographic areas (ie, the minimum distance from VE4 and GU1 to their closest populations is about 30 km). Both populations are found within isolated mountain valleys surrounded by dense forest (GU1) or dense forest and sea (VE4). Interconnection with other populations therefore seems highly limited. A second factor acting on diversity is the temporal stability of habitats. For example, VE2, VE4, GU1 and BR1 experience strong variation in water availability, including complete drying out. The end of the dry season/beginning of the rainy season is likely to be associated with strong reduction in population size. Population dynamics studies have indeed documented such variation, and even population extinction in B. glabrata (eg, St. Lucia, Sturrock, 1973; Puerto Rico, Jobin, 1970; Guadeloupe, figure 3 in Pointier et al, 1991b; Venezuela, figure 4 in Pointier et al, 1991a), as well as in other tropical snails (Charbonnel, 2001). The effective size of these populations is expected to be low, which may explain the limited diversity (Crow and Kimura, 1970).
Mating system
Our results from tests of both Hardy-Weinberg equilibrium and genotypic disequilibrium indicate that outcrossing is the major mating system in most populations studied, with a mean selfing rate of 0.05. This is consistent with previous studies in the same species (Mulvey and Vrijenhoek, 1982; Vianey-Liaud, 1997; Mavárez et al, 2000, 2002a). Moreover, this reasonably fits the bimodal distribution of selfing rates observed in freshwater snails, with most species being either preferential selfers or preferential outcrossers (see review in Städler and Jarne, 1997; and also Viard et al, 1997a, b; Mavárez et al, 2000a, 2002a; Meunier et al, 2001; Charbonnel et al, 2002; this study). Note that the association of predominant outcrossing and high level of genetic diversity still reinforces the observed correlation between mating systems and diversity observed in plants (Hamrick and Godt, 1989; Awadalla and Ritland, 1997) and snails (Jarne, 1995).
Is B. glabrata a single genetic entity?
A striking pattern was uncovered in the VE6 and VE13 populations, both made of two groups, one of outcrossing individuals and the second of the same multilocus genotype (extremely divergent from other genotypes). These two groups appears also highly divergent in regards to ITS-2 and 16S gene sequences (Mavárez et al, 2002b) We envisaged two explanations. (i) B. glabrata uncovers a species-complex, and two species coexist within VE6 and VE13. One of these is characterised by a single, invariable multilocus microsatellite genotype. The lack of genetic exchange between the two species is attested by the fact that the alleles of this genotype are not found elsewhere (with few exceptions). Note that the mixing of cryptic species is easily detected because one of them lacks intra-specific genetic variation. However we can be quite confident that cryptic species do not occur elsewhere (at least in northern South America) because linkage disequilibria are significant only in localities VE6 and VE13. The possibility of species- complex in B. glabrata has already been suggested (see Woodruff and Mulvey, 1997 and references therein; Mavárez et al, 2002b). However, they were never formally identified. (ii) These populations have been recently invaded by a genotype that strongly self-fertilises. Such a shift in the mating system (from cross- to self-fertilisation) has not been previously reported in animals, but there is some evidence in plants (Husband and Barrett, 1991; Liu et al, 1999). Selfing precludes gene flow from the rest of B. glabrata populations. However, even highly selfing species usually practise a bit of outcrossing, and recombination between genotypes eventually occurs. Moreover, even if all ovules are self-fertilised, there is no reason why individuals would not transfer sperm to outcrossers by copulating as males. Therefore, the lack of recombinant genotypes would, in our case, mean that the selfing variant has invaded both populations quite recently. However, recombinants may be eventually found given time and sampling effort. More taxonomic work is required here, as well as detailed analysis of the mating behaviour and mating system (see Ritland, 1986; Viard et al, 1997a).
References
Awadalla, P, Ritland, K (1997). Microsatellite variation and evolution in the Mimulus guttatus species complex with contrasting mating systems. Mol Biol Evol 14: 1023–1034.
Barton, NH, Whitlock, MC (1997). The evolution of metapopulations. In: Hanski I, Gilpin M (eds) Metapopulation Biology: Ecology, Genetics and Evolution Academic Press: London, pp 183–210.
Belkhir, K, Borsa, P, Chikhi, L, Raufaste, N, Bonhomme, F (2000). GENETIX 4. 02, logiciel sous Windows TM pour la génétique des populations. Université de Montpellier II, Montpellier (France).
Brown, DS (1994). Freshwater Snails of Africa and Their Medical Importance. Taylor and Francis Ltd: London.
Charbonnel, N (2001). De la Génétique à la Dynamique des Populations: l’Example du Gastéropode des Eaux Douces, Biomphalaria pfeifferi, à Madagascar. PhD Thesis, Ecole Nationale Supérieure Agronomique de Montpellier, Montpellier.
Charbonnel, N, Angers, B, Debain, C, Razatavonjizay, R, Bremond, P, Jarne, P (2002). Fine scale genetic and demographic studies reveal complex metapopulation dynamics in Biomphalaria pfeifferi, the intermediate host of Schistosoma mansoni. J Evol Biol 15: 248–261.
Charbonnel, N, Angers, B, Debain, C, Razatavonjizay, R, Bremond, P, Jarne, P The influence of mating system, demography, parasites and colonisation on the genetic structure of Biomphalaria pfeifferi in Madagascar. Mol Ecol. (submitted).
Charlesworth, B, Morgan, MT, Charlesworth, D (1993). The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303.
Crow, JF, Kimura, M (1970). An Introduction to Population Genetics Theory. Harper and Row: New York.
Excoffier, L, Smouse, PE, Quattro, JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Freeland, JR, Romualdi, C, Okamura, B (2000). Gene flow and genetic diversity: a comparison of freshwater bryozoan populations in Europe and North America. Heredity 85: 498–508.
Giles, BE, Goudet, J (1997). Genetic differentiation in Silene dioica metapopulations: Estimation of spatiotemporal effects in a successional plant species. Am Nat 149: 507–526.
Goudet, J (2001). FSTAT, a program to estimate and test gene diversities and fixation indices (version 293). Available at http://www.unil.ch/izea/softwares/fstat.html.
Goudet, J, Raymond, M, de Mees, T, Rousset, F (1996). Testing differentiation in diploid populations. Genetics 144: 1933–1940.
Hamrick, JL, Godt, JW (1989). Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kalher AL, Weir BS (eds) Plant Population Genetics, Breeding, and Genetic Resources Sinauer Associates Inc: Sunderland, pp 43–63.
Harrison, S, Hastings, A (1996). Genetic and evolutionary consequences of metapopulation structure. Trends Ecol Evol 11: 180–183.
Husband, BC, Barrett, SCH (1991). Colonization history and population genetic structure of Eichhornia paniculata in Jamaica. Heredity 66: 287–296.
Jarne, P (1995). Mating system, bottlenecks and genetic polymorphism in hermaphroditic animals. Gen Res 65: 193–207.
Jarne, P, Delay, B (1991). Population genetics of freshwater snails. Trends Ecol Evol 6: 383–386.
Jarne, P, Vianey-Liaud, M, Delay, B (1993). Selfing and outcrossing in hermaphrodite freshwater gastropods (Basommatophora): where, when and why. Biol J Linn Soc 49: 99–125.
Jobin, WR (1970). Population dynamics of aquatic snails in three farm ponds of Puerto Rico. Am J Trop Med Hyg 19: 1038–1048.
Jones, CS, Lockyer, AE, Rollinson, D, Piertney, SB, Noble, LR (1999). Isolation and characterization of microsatellite loci in the freshwater gastropod, Biomphalaria glabrata, an intermediate host for Schistosoma mansoni. Mol Ecol 8: 2149–2151.
Liu, F, Charlesworth, D, Kreitman, M (1999). The effect of mating system differences on nucleotide diversity at the Phosphoglucose Isomerase locus in the plant genus Leavenworthia. Genetics 151: 343–357.
Madsen, H, Frandsen, F (1989). The spread of freshwater snails including those of medical and veterinary importance. Acta Trop 46: 139–146.
Mavárez, J, Amarista, M, Pointier, J-P, Jarne, P (2000). Microsatellite variation in the freshwater schistosome-transmitting snail Biomphalaria glabrata. Mol Ecol 9: 1009–1011.
Mavárez, J, Amarista, M, Pointier, J-P, Jarne, P (2002a). Fine-scale population structure and dispersal in Biomphalaria glabrata, the intermediate snail host of Schistosoma mansoni, in Venezuela. Mol Ecol 11: 881–892.
Mavárez, J, Steiner, C, Pointier, J-P, Lenormand, T, Jarne, P (2002b). Evolutionary history and phylogeography of the schistosome-transmitting freshwater snail Biomphalaria glabrata based on nuclear and mitochondrial DNA sequences. Heredity 89: 266–272.
McCauley, DE (1991). Genetic consequences of local population extinction and recolonization. Trends Ecol Evol 6: 5–8.
Meunier, C, Tirard, C, Hurtrez-Boussès, S, Durand, P, Bargues, MD, Mas-Coma, S et al. (2001). Lack of molluscan host diversity and the transmission of an emerging parasitic disease in Bolivia. Mol Ecol 10: 1333–1340.
Mulvey, M, Newman, MC, Woodruff, DS (1988). Genetic differentiation among West Indian populations of the schistosome-transmitting snail Biomphalaria glabrata. Malacologia 29: 309–317.
Mulvey, M, Vrijenhoek, RC (1982). Population structure in Biomphalaria glabrata: examination of an hypothesis for the patchy distribution of susceptibility to schistososmes. Am J Trop Med Hyg 31: 1195–1200.
Nei, M (1987). Molecular Evolutionary Genetics. Columbia University Press, New York.
Pálsson, S (2000). Microsatellite variation in Daphnia pulex from both sides of the baltic sea. Mol Ecol 9: 1075–1088.
Paraense, WL (2001). The schistosome vectors in the Americas. Mem Inst Osw Cruz 96: 7–16.
Petit, RJ, El Mousadik, A, Pons, O (1998). Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12: 844–855.
Pointier, J-P (1976). Répartition locale et biogéographie des mollusques dulçaquicoles de la Guadeloupe (Antilles françaises). Malac Rev 9: 85–103.
Pointier, J-P (1999). Invading freshwater gastropods: some conflicting aspects for public health. Malacologia 41: 403–411.
Pointier, J-P, Balzán, C, Chrosciechowski, P, Incani, RN (1991a). Limiting factors in biological control of the snail intermediate hosts of Schistosoma mansoni in Venezuela. J Med Appl Malac 3: 53–67.
Pointier, J-P, Theron, A (1979). Intestinal schistosomiasis in the freshwater mangrove of Guadalupe (French Antilles). Ecology of the vector, Biomphalaria glabrata and its parasite Schistosoma mansoni. Ann Parasit Hum Comp 54: 43–56.
Pointier, J-P, Théron, A, Borel, G (1993). Ecology of the introduced snail Melanoides tuberculata (Gastropoda: Thiaridae) in relation to Biomphalaria glabrata in the marshy forest zone of Guadeloupe, French West Indies. J Moll Stud 59: 421–428.
Pointier, J-P, Théron, A, Imbert-Establet, D, Borel, G (1991b). Eradication of a sylvatic focus of Schistosoma mansoni using biological control by competitor snails. Biol Cont 1: 244–247.
Raymond, M, Rousset, F (1995a). An exact test for population differentiation. Evolution 49: 1280–1283.
Raymond, M, Rousset, F (1995b). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Ritland, K (1986). Joint maximum likelihood estimation of genetic and mating structure using open-pollinated progenies. Biometrics 42: 25–43.
Rousset, F (1996). Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142: 1357–1362.
Rousset, F (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Rousset, F (2001). Genetic approaches to the estimation of dispersal rates. In: Clobert J, Danchin E, Dhondt AA, Nichols J (eds) Dispersal. Oxford University Press: New York, pp 18–28.
Rousset, F, Raymond, M (1995). Testing heterozygote excess and deficiency. Genetics 140: 1413–1419.
Schneider, S, Kuffer, JM, Roessli, D, Excoffier, L (1999). Arlequin Version 11: A software for population genetic data analysis. University of Geneva, Switzerland.
Sire, C, Durand, P, Pointier, J-P, Theron, A (1999). Genetic diversity and recruitment pattern of Schistosoma mansoni in a Biomphalaria glabrata snail population: a field study using RAPD markers. J Parasitol 85: 436–441.
Sokal, RR, Rohlf, FJ (1995). Biometry. Freeman and Company, New York.
Städler, T, Jarne, P (1997). Population biology, genetic structure, and mating system parameters in freshwater snails. In: Streit B, Städler T, Lively CM (eds) Evolutionary Ecology of Freshwater Animals Birkhäuser Verlag: Basel, pp 231–262.
Sturrock, RF (1973). Field studies on the transmission of Schistosoma mansoni and on the bionomics of its intermediate host, Biomphalaria glabrata, on St. Lucia, West Indies. Int J Parasit 3: 175–194.
Vianey-Liaud, M (1997). La reproduction chez un mollusque hermaphrodite simultané, la planorbe Biomphalaria glabrata (Say, 1818) (Gastéropode, Pulmoné). Haliotis 27: 67–114.
Viard, F, Doums, C, Jarne, P (1997a). Selfing, sexual polymorphism and microsatellites in the hermaphroditic freshwater snail Bulinus truncatus. Proc Roy Soc Lond B 264: 39–44.
Viard, F, Justy, F, Jarne, P (1997b). The influence of self-fertilization and bottlenecks on the genetic structure of subdivided populations: a case study using microsatellite markers in the freshwater snail Bulinus truncatus. Evolution 51: 1518–1528.
Wade, MJ, McCauley, DE (1988). Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution 42: 995–1005.
Weir, BS, Cockerham, CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whitlock, MC (1992). Nonequilibrium population structure in forked fungus beetles: extinction, colonization and the genetic variance among populations. Am Nat 139: 952–970.
Woodruff, DS, Mulvey, M (1997). Neotropical schistosomiasis: African affinities of the host snail Biomphalaria glabrata (Gastropoda: Planorbidae). Biol J Linn Soc 60: 505–516.
Acknowledgements
This work was funded by the Ecos-Nord Committee V99A02 and the Programme National Dynamique de la Biodiversité et Environnement (to PJ). JM benefits from a fellowship from the Consejo Nacional de Investigaciones Científicas y Tecnológicas (Conicit), Venezuela. We would like to thank Mr Manuel Amarista at the Instituto de Altos Estudios en Salud Pública Dr Arnoldo Gabaldón, Maracay, for his collaboration during field trips to Venezuela and Mr Alexis Guevara for his help to collect snails. We also thank Mrs C Debain for technical assistance and Dr N Charbonnel for helpful revision of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mavárez, J., Pointier, JP., David, P. et al. Genetic differentiation, dispersal and mating system in the schistosome-transmitting freshwater snail Biomphalaria glabrata. Heredity 89, 258–265 (2002). https://doi.org/10.1038/sj.hdy.6800127
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6800127
Keywords
This article is cited by
-
The population genetic structure of Biomphalaria choanomphala in Lake Victoria, East Africa: implications for schistosomiasis transmission
Parasites & Vectors (2014)
-
Meta-analysis indicates lack of local adaptation of Schistosoma mansoni to Biomphalaria alexandrina in Egypt
Parasitology Research (2014)
-
Transgenerational cross-tolerance to stress: parental exposure to predators increases offspring contaminant tolerance
Ecotoxicology (2013)
-
Transmission dynamics of two strains of Schistosoma mansoni utilizing novel intermediate and definitive hosts
Parasitology Research (2011)
-
Evolutionary history and phylogeography of the schistosome-vector freshwater snail Biomphalaria glabrata based on nuclear and mitochondrial DNA sequences
Heredity (2002)
