
Abstract
Matrilineal phylogenetic divergence among Atlantic salmon stocks of the Bay of Fundy in south eastern Canada is investigated. Sequence variation in two regions of the mitochondrial ND1 gene, encompassing 710 base pairs, is described for 168 salmon from 11 rivers. Mean overall haplotype and nucleotide diversity (h and π) observed are 0.5014 and 0.00095, respectively. Nested clade analysis (NCA) and molecular analysis of variance (AMOVA) both point to highly restricted gene flow among rivers and show the haplotype distribution to be geographically structured. Variation among predefined regions of the Bay (16%) is greater than among populations within these regions (14%) The main regional differentiation occurs between rivers of the geographically isolated inner Minas Basin and those elsewhere in the Bay. Differentiation most probably reflects the pattern and nature of the historical processes associated with post-glacial colonisation of the area by salmon following the last Pleistocene glacial maximum c. 18000 yrs BP.
Similar content being viewed by others

Introduction
Nearctic and Paleartic species such as Atlantic salmon (Salmo salar L.) have experienced dramatic historical range shifts due to the advance and retreat of Pleistocene ice sheets and the associated climate change. The species’ modern North American range, and much of its distribution in Europe, was covered by ice at the time of the last glacial maximum (LGM) (MacCrimmon and Gots, 1979; Benn and Evans, 1998). Thus historical factors associated with the post-glacial establishment of modern populations will be major determinants of the species’ phylogeography (Bernatchez and Wilson, 1995; Hewitt, 2000). At least among established populations, contemporary gene flow is likely to be severely restricted as tagging studies show a strong homing to natal streams to spawn (Stabell, 1984; Youngson et al, 1994). This is consistent with the observed widespread genetic differentiation of populations both between and within rivers (Verspoor, 1997).
What is poorly understood for most Nearctic and Palearctic species, such as Atlantic salmon, is whether they have evolved distinct phylogeographic lineages as part of the post-glacial recolonisation process. Taxonomists currently view the Atlantic salmon as monophyletic, with the last subdivision of the species, into anadromous (S. s. salar) and non-anadromous (S. s. sebago) forms, rejected on the basis of morphology (Wilder, 1947). The absence of a phylogenetic distinction between the two life-history forms is supported by allozyme studies in both Europe and North America (Ståhl 1987; Verspoor, 1994) though anadromous and non-anadromous populations have diverged sufficiently in some cases to exist sympatrically (Verspoor and Cole, 1989). However, molecular studies do show a deep phylogenetic division between North American and European salmon stocks (Ståhl 1987; Verspoor, 1997; King et al, 2000), and a shallower division of European stocks into Baltic and Atlantic (ie, non-Baltic) groups (Ståhl, 1987; Koljonen et al, 1999; Verspoor et al, 1999; Nilsson et al, 2001). In North America, regional differences have been detected in relation to transferrin variation (Verspoor, 1986) but the evolutionary basis of these regional differences is unclear and detailed phylogeographic studies of North American populations are lacking.
Phylogenetic divergence of Baltic and non-Baltic European salmon is associated with phenotypic differences in migratory behaviour; marine migration of Baltic stocks being confined to the Baltic Sea while other European stocks utilise the Atlantic Ocean (Mills, 1989). Major regional differentiation in migratory behaviour, as well as other life-history traits, has also been reported for salmon stocks of the Bay of Fundy in south eastern Canada. Recoveries of tagged salmon from inner Bay (iBoF) rivers have been largely restricted to the Bay or immediately adjacent sectors of the Gulf of Maine (Jessop, 1976) while salmon tagged elsewhere in North America are typically recovered in the seas off Newfoundland, Labrador and Greenland (Mills, 1989). Furthermore, returning adults in iBoF stocks almost all first mature after only one winter at sea (ie grilse) as well as being iteroparous (Ducharme, 1969) whereas in most other anadromous North American stocks, a large proportion of fish mature after 2 or more years at sea, and most fish spawn only once (Mills, 1989).
Bay of Fundy salmon stocks are perilously close to extinction (DFO, 1999; P Amiro, unpublished) and it is crucial to establish if life history differentiation within the Bay of Fundy has a phylogeographic association, as observed for Baltic and non-Baltic stocks in Europe. The stocks with the different life-history traits should be managed independently to ensure the continued survival of different unique components of the species’ intraspecific biodiversity. The study of mitochondrial DNA (mtDNA) differentiation among Atlantic salmon stocks in the Bay of Fundy, described here, was carried out to address this question and to advance general understanding of the importance of post-glacial colonisation processes as determinants of the patterns and levels of intraspecific biodiversity in northern species.
Materials and methods
Samples
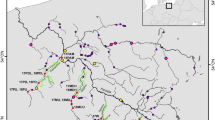
Skin or fin tissue were obtained for genetic typing from 168 juvenile Atlantic salmon from 11 rivers (Figure 1; Table 1). These rivers span the coastline of the Bay of Fundy in eastern North America from the Stewiake River, at the head of the Minas Basin in the Inner Bay, to the Narraguagus River where the Bay becomes the Gulf of Maine. The samples, with the exception of the Narraguagus, Gaspereau and Saint John rivers, were composed of juvenile fish of mixed age class caught in 1999 by electro-fishing and sampled non-destructively. The Narraguagus sample was collected from the river in 1996. The Gaspereau fish are first generation hatchery offspring from crosses made from 10 and 11 wild-caught females and males, respectively, spawned in 1998. The Saint John sample are offspring of over 300 wild fish spawned in 1992. All tissues were stored in absolute ethanol at the time of collection.

Rivers in Bay of Fundy region in eastern North America with historical sports catches of Salmo salar. Those in white were found to be devoid of salmon juveniles in 2000 while those with a ? were not surveyed but are suspected to have only remnant populations. The rivers enclosed by the thin solid line are considered to be inner Bay rivers based on life-history and behavioural characteristics. Numbered rivers (Table 1) were sampled for the current study. The dashed line shows the ice edge c. 15000 years bp and the hatching the area which would at the time have been below sea level based on 27 Shaw et al, (2002) and J Shaw (personal communication).
Genetic typing
Sequence analysis was carried out on DNA extracted following the procedure described by Knox et al (2002). Two sub regions of 350 and 360 base pairs of the mtDNA ND1 gene were sequenced with an ABI 377 DNA analyser following the manufacturers instructions, using both forward and reverse cycle sequencing based on PCR primer sets 1 and 2 of Knox et al (2002). The primers are nested within a larger gene region analysed for restriction fragment length polymorphism (RFLP) variation by Verspoor et al (1999). The sequence data encompasses base positions 3666 to 4015 and 4141 to 4500 of Hurst et al (1999; Genbank Accession No. U12143) which contain previously reported polymorphic restriction sites (Verspoor et al, 1999).
Statistical analysis
A molecular analysis of variance (AMOVA, Version 1.5; Excoffier et al, 1992) was carried out with regard to three regions, Minas Basin, Chignecto Bay, and outer Bay of Fundy, defined a priori on the basis of geography and population. The Minas basin represents a geographically isolated inner region of the Bay of Fundy (Figure 1). The division of the remaining part of the study areas, into Chignecto Bay and outer Bay of Fundy sites, is based on the differentiation between the salmon stocks in these two adjacent regions with regard to life history and marine migration (Ducharne, 1969; Jessop, 1976); the stocks in the Chignecto Bay group have a limited marine migration and a high incidence of iteroparity while those in the outer group have an extensive marine migration and limited iteroparity. Divergence among haplotypes was measured as squared Euclidian distance. Pairwise ΦST estimates were calculated and tested for significance based on 1000 permutations.
Nested clade analysis (NCA – Templeton et al, 1995; Templeton, 1998) was implemented using GeoDis (Posada et al, 2000) based on a nested cladogram for haplotype variation constructed according to recommended procedures (Templeton et al, 1992; Templeton and Sing, 1993). The analysis used a matrix of relative geographic distance based on shortest marine distance between river mouths. Estimates of nucleotide divergence among haplotypes derived using MEGA 2.1 (Kumar et al, 2001) were used to generate population estimates for haplotype and nucleotide divergence using REAP (McElroy et al, 1991).
Results
Diversity analysis
Sequence analysis shows seven variable sites within the two regions analysed. These define 10 haplotypes, each separated from its most closely related haplotypes by a single mutation (Figure 2). The minimum spanning network (Figure 2) has unambiguous linkages among haplotypes with the exception of haplotype vii, which could equally be derived from haplotypes vi and viii. The network is most parsimoniously linked to the European haplotype group through haplotype i; there are only two main salmon lineages – North American and European (King et al, 2000; Nilsson et al, 2001). Five mutations (0.7% divergence) in the sequenced regions separate the two haplotypes based on the data of Hurst et al (1999; GenBank U12143); six or more mutations separate this European variant from the other haplotypes detected in the present study. Linkage to the European lineage through haplotype i is consistent with the position of this haplotype at the centre of the radiation within the North American network.

The minimum spanning network for haplotype variation detected in Atlantic salmon stocks in Bay of Fundy rivers by direct sequence analysis of 349 bp and 361 bp regions of the gene. Base changes leading to amino acid substitutions in the ND1 protein are indicated. Haplotypes refer to those in Table 1. E stands for the European haplotype of Hurst et al (1999). The two possible clade nesting arrangements for NCA, taking into account the ambiguity in the linkage of haplotype vii, are shown.
Haplotype and nucleotide diversity (h and π) in the two inner Bay regions (Table 1) were not statistically different (Mann-Whitney U test for h and π both non-significant) but, in a pooled comparison, show significantly higher values than the outer Bay region (Mann-Whitney U test for h: P = 0.05 and π: P = 0.05). No single haplotype was observed in all 11 river samples. Haplotype i, detected in eight rivers, was the most common variant and represented 40% of individuals; 30% of fish sampled were haplotype viii, the second most common and most widespread variant, found in nine of the 11 samples (Table 1). The third most common variant, haplotype ix, was restricted to the six river samples from the Minas Basin (Figure 1; Table 1). These three common haplotypes are all interior variants in the network (Figure 2) while five of the seven remaining variants are tip haplotypes and restricted to single samples with three observed in only a single individual. The remaining two tip haplotypes, v and vi, occur in multiple individuals in three geographically adjacent rivers.
Variation within and among populations
Significant variation among regions and populations within regions is revealed by the AMOVA (Table 2). Including molecular distance data, regional and population differences represent 16% and 14% of the total variance, respectively. Over half of pairwise tests under AMOVA of divergence among samples (Table 3) are significant at the 0.001 level (33/55) with a smaller proportion of comparisons within regions (7/19) than among regions (26/36) being significant (P = 0.012 – Fisher’s exact test). The two rivers of the outer Bay region, widely separated geographically, show no significant difference. However, paired regional comparisons using AMOVA show significance of the regional variance component in the overall analysis to be largely attributable to divergence of the Minas Basin region; of the paired comparisons only the test comparing the Chignecto Bay and Outer Bay populations was non-significant (P = 0.3556).
NCA also indicates variation among samples is associated with geography. For the analysis, the two alternative clade nesting arrangements, generated by the ambiguity of the clustering of haplotype vii, were used (Figure 2). Arrangement A, with vii linked to viii, assumes transitions are more likely than transversions while B, with vii linked to vi, is spatially conservative given these two haplotypes were detected in the same sample; type viii is absent from the sample with type vii. With both nesting arrangements, the results of the contingency analysis (Table 4) show significantly non-random distributions within the main lower level clade as well as within the higher clades. However, in arrangement A, lower clade 1–2, which includes the single haplotype vii individual, shows a near significant result. This arises because in the sample which contains the one type vii individual, this is the only representative of clade 1–2. As the other type in this clade in arrangement A (ie viii) is widespread and abundant, this is unexpected under a model that the distribution of the two types within the clade is random. In contrast, in the geographically conservative arrangement B, the distribution of vii within the new clade 1–2 is not unexpected. This difference aside, the two arrangements show the same non-random association of variation with geography.
Insight into the nature of the non-random association is given by the distance analysis within the NCA. For the more geographically conservative nesting arrangement B, the average distance of individuals of a given clade from the geographical centre of distribution of that clade, Dc, are found to be significantly small in many cases. This is true for two tip clades at the lowest nesting level – haplotypes iv and v with clade 1–1, and for all tip clades at higher levels. Thus the distribution of individuals within these clades is more spatially restricted than expected (Figure 3). In contrast, with the exception of the lowest level, all interior clades are significantly more widespread than expected. Furthermore, within all clades where significantly small Dc values occur for tip clades, the differences between interior and tip clades, ie (Int-Tip)c distances, are significantly large as are the (Int-Tip)n distances in the higher level clades.

Results of nested clade analysis. Haplotype designations are those in Table 1 and Figure 2. Boxing shows nesting of clades as in arrangement B in Figure 2. Dc refers to the average distance of individuals in a clade from the geographical centre of the distribution of all individuals of that clade observed in all samples. Dn refers to the average distance of individuals in a clade from the geographical centre of all individuals in all clades associated with the next step clade within which the clade in question is nested. The S and SS, and L and LL, superscripts refer to significantly small and large values at the 0.05 and 0.01 levels, respectively. (Int-Tip)c and (Int-Tip)n values at the bottom of each box give the average differences in distances, and their significance, between interior and tip clades within the nested group for Dc and Dn, respectively. Interior clades are identified by shading. Significance of values is based on permutation analysis using 1000 resamples.
The occurrence of significant Dc, Dn and I-T values argues for the rejection of the null hypothesis of no geographical association of variation and, based on the inference key of Templeton et al (1995), supports restricted gene flow with isolation by distance (Step 4). Dc values are in all cases smaller for tip than for interior clades, at all clade levels and tend to increase with increasing clade level (Figure 3), as expected if tip clades are younger than interior clades and have less opportunity to become widely distributed compared to linked interior clades (Castelloe and Templeton, 1994). At the same time, there are no reversals of Dn and (Interior-Tip)n compared to Dc values, no tip clades have significantly large Dn values, and no Dn and (Interior-Tip) values for interior clades are significantly small.
Discussion
More variation was uncovered in the ND1 gene region by sequencing than would have been resolved by RFLP analysis. Of the seven variable sites, only variation at site 4199 would be detectable by restriction enzyme (Alu I). Even so, the level of variation observed is higher than expected. Only two variable sites were reported for the same part of the gene in 32 salmon derived from a much larger geographical area spanning both Baltic and non-Baltic phylogenetic groups in Europe (Nilsson et al, 2001). Haplotype (h) and nucleotide diversity (π) observed for Baltic and non-Baltic salmon for the gene region studied were 0.3282 (±0.0768) and 0.000475, and 0.5362 (±0.0904) and 0.000975, respectively (based on data in Nilsson et al, 2001). Average Bay of Fundy values for h and π (Table 1) are similar to the more diverse non-Baltic European salmon. Unexpectedly, inner Bay rivers show significantly more genetic variability than those of the outer Bay. Angling statistics and catchment size suggest the historical population of the Saint John River, in the outer Bay, would have been several fold greater than any of the inner Bay rivers and that the latter populations have been severely depressed in recent years (DFO, 1999; P Amiro, unpublished).
Both AMOVA and NCA show significant heterogeneity in haplotype frequencies among rivers which, given the high diversity within samples, is unlikely to be an artefact of sampling restricted numbers of families within rivers. Furthermore, the heterogeneity is regionally distributed, supporting a hypothesis of historically restricted gene flow among rivers within the Bay. This conclusion is consistent with observed differentiation between two of the rivers at microsatellite loci (McConnell et al, 1997) and general evidence that straying of anadromous Atlantic salmon from their natal streams is very limited (Mills, 1989; Youngson et al, 1994). However, the observation that variation among regions is greater than within regions is surprising as significant regional differentiation has only previously been reported on larger geographical scales. The division of European stocks into Baltic and non-Baltic groups (Koljonen et al, 1999; Nilsson et al, 2001) involves a spatial scale where the Baltic Sea alone, at 422 000 km2, is over 20-fold larger than the Bay of Fundy.
The regional differentiation observed is predominantly attributed to the difference between populations from the geographically distinct Minas Basin rivers and those elsewhere in the Bay of Fundy. At the same time, levels of differentiation among populations within the Basin are low compared to levels among populations elsewhere in the Bay (Table 3). The regional differentiation is due largely to >35% of Basin fish being of haplotype ix, a variant not detected elsewhere in the study area (Table 1). Interestingly, clade 1–3 individuals, which include haplotypes ix and x and are detectable by RFLP analysis of the ND1 gene using Alu I, have not been observed elsewhere in North America (Tessier et al, 1997; King et al, 2000; Verspoor, unpublished). Collectively, this strongly supports stocks in the Minas Basin having a unique evolutionary history, at least on the maternal side.
Whether further regional differentiation is present is unclear. The analysis suggests that the rivers of Upper Chignecto Bay could also be distinctive both from Minas Basin and outer Bay rivers. Despite being the second most common haplotype, viii was only observed in one of 25 fish in the Saint John and the Narraguagus, the two outer Bay rivers sampled. These two rivers show <30% of the differentiation observed between the Saint John River and its nearest neighbour, the Black river (Figure 1; Table 3) yet the Saint John and Narraguagus are separated by the largest sampling gap in the study (Figure 1). Further regional differentiation is also suggested by the NCA. Haplotypes v and clade 1–2, which are confined to the Upper Chignecto Bay stocks, have significantly constrained distributions. However, regional division of populations in the study area outside the Minas Basin is not supported by the AMOVA which may reflect the small number of rivers analysed from the Chignecto and outer Bay areas. Further sampling is needed to clarify this point.
Both AMOVA and NCA support a phylogenetic basis to regional differentiation. The inference key of Templeton et al (1995) for the NCA points to regional differentiation caused by restricted gene flow with isolation-by-distance (IBD) rather than past fragmentation of the distribution. This seems unlikely to be simply a reflection of contemporary gene flow. Almost all rivers in the inner Bay region still containing substantive salmon populations were sampled (Figure 1). Some range fragmentation has occurred in the recent past, with the loss of salmon stocks in rivers from some parts of the inner Bay eg, the upper Chignecto Bay. However, the Minas basin rivers are geographically as close to the sampled rivers in the upper Chignecto Bay as to any extinct or residual populations not sampled (Figure 1). Furthermore, historical gene flow within the Bay of Fundy as a whole has most likely been a function of straight-line geographic separation rather than simply coastal distance. Gradual change with distance, rather than sharp discontinuities, would be expected if a model of IBD involving contemporary gene flow was responsible for the observed differentiation.
Historical conditions in the study area suggest the observed discontinuities are likely to reflect the manner in which the Bay of Fundy was recolonised after the last glacial maximum (LGM) c. 18000 years BP when the entire area was covered by ice sheets (Pielou, 1991). Reconstruction of ice cover in the Fundy region (Figure 1) suggests that c. 15000 yrs BP one small spatially distinct area along the ice edge may have been the first to have had non-glacial rivers with suitable salmon habitat. This area, known as the Caledonia and Kent hills, are c. 400 m above sea level and drained by the small coastal streams of the upper Chignecto Bay including the Big Salmon, Black and Irish Rivers. At this time the modern coastal area to the south-west was mostly below sea level and adjacent rivers would still have drained the main ice sheet while areas to the north and east, such as the Minas Basin, were still covered by ice (Figure 1). The view that the inner Bay was the first part of the study area to be colonised fits with the higher genetic diversity observed there compared to the rivers of the outer Bay, given the general view that areas colonised first show higher genetic diversity than those colonised later (Avise, 2000).
Upper Chignecto Bay rivers would very likely already have had salmon populations at this early stage judging by the modern situation in West Greenland. In West Greenland, one self-sustaining Atlantic salmon population occurs naturally near the ice edge in the Kapisidlit River, a small, non-glacial coastal stream c. 10 km long at the head of a fjord c. 8 km from the main ice sheet (Nielson, 1961). This river is similar in size to some of the smaller Upper Chignecto rivers. Interestingly, during periods with favourable climatic conditions, strays from the Kapisidlit River establish ephemeral populations in other small, neighbouring non-glacial rivers which go extinct when conditions worsen. With gradually improving conditions, it could be envisaged that this metapopulation dynamic would be the main force driving colonisation in areas such as the Bay of Fundy, as the area deglaciated, once the first population(s) had been established by colonisers from the main Pleistocene refugia. The latter would have been hundreds, if not thousands, of kilometres to the south in rivers along the coast to the south of modern-day New York City, or possibly, in rivers in present-day marine areas such as the George’s Bank which was above sea-level and unglaciated at the time (Pielou, 1991).
If straying among local rivers was more common than long distance migration, colonisation within the Bay would have been driven largely by fish straying from the upper Chignecto Bay area to other small, potentially isolated coastal areas, in the Bay. One of these would have been the Minas basin, as rivers in these areas became habitable. The reconstruction of the post-glacial history of Atlantic Canada (Shaw et al, 2002) suggests the history of the Minas Basin may have had a particular twist which promoted development of the regional differentiation observed. This shows that for several thousand years following glacial retreat all the rivers in the Basin may have been tributaries of a single river/lake system. The mouth of this river system appears to have been near Cape Split (Figure 1), the headland which currently separates the Basin from the rest of the Bay.
The historical processes driving differentiation would under this scenario have been dominated by founder events, initially associated with long distance colonisation into the region from Pleistocene refuges and, later, with migration among rivers within the Bay. Founding variants, and newly arising local mutations, could then have become regionally common through genetic drift and the local range expansion described. As each area expanded in size and included more rivers, these would again be most likely to have populations founded from neighbouring rivers with existing populations, until the more or less continuous distribution seen in the Bay of Fundy, in historical times, emerged. The resulting differentiation among and within regions of the Bay would then have been maintained by natal homing behaviour. Straying was probably always only a sporadic and only occasionally successful event. All salmonids show strong natal homing (Stabell, 1984) and the trait in Atlantic salmon is most likely ancient. The lack of success of strays would have become more acute to the extent that populations became locally adapted and acted to maintain the pattern of differentiation emerging from the colonisation process.
The exact nature of the historical processes responsible for the observed regional differentiation is still a matter for speculation. However, what is clear is gene flow between salmon populations in the Minas Basin and those in other parts of the Bay of Fundy has been restricted for much of the time since the Basin was first colonised. This strongly supports the view that Basin populations represent a distinct evolutionary grouping. Whether the same is true for Chignecto Bay and Outer Bay stocks, and whether a phylogenetic split between inner and outer Bay rivers underlies their general life-history divergence, remains to be established. The possibility cannot be dismissed. Studies show an abundance of intraspecific genetic diversity in salmonid fishes, including Atlantic salmon, manifested in ecological and behavioural specialisation not associated with obvious morphological divergence (Behnke, 1972). Furthermore, natural selection associated with the invasion of novel habitats, with new and evolving environmental conditions, appears to be linked to adaptive evolutionary divergence (Orr and Smith, 1998). This suggests that Atlantic salmon stocks, in areas previously covered by Pleistocene glaciers, may well represent a mosaic of phylogeographic groups, each a unique, locally adapted component of the species’ intraspecific variation.
References
Avise, JC (2000). Phylogeography: the history and formation of species. Harvard University Press: Cambridge, Mass., USA.
Behnke, RJ (1972). The systematics of salmonid fishes of recently glaciated lakes. J Fish Res Bd Canada, 29: 639–671.
Benn, DI, Evans, DJA (1998). Glaciers and Glaciation. Arnold: London.
Bernatchez, L, Wilson, CC (1998). Comparative phylogeography of Nearctic and Paleartic fishes. Mol Ecol, 7: 431–452.
Castelloe, J, Templeton, AR (1994). Root probabilities for intraspecific gene trees under neutral coalescent theory. Mol Phylog Evol, 3: 102–113.
DFO (1999). Interaction between wild and farmed Atlantic salmon in the Maritime Provinces. DFO Maritimes Regional Habitat Status Report, 99/1E.
Ducharme, LJA (1969). Atlantic salmon returning for their fifth and sixth consecutive spawning trips. J Fish Res Bd Canada, 26: 1661–1664.
Excoffier, L, Smouse, PE, Quattro, JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131: 479–491.
Hewitt, G (2000). The genetic legacy of the Quaternary ice ages. Nature, 405: 907–913.
Hurst, CD, Bartlett, SE, Davidson, WS, Bruce, IJ (1999). The complete mitochondrial DNA sequence of the Atlantic salmon, Salmo salar. Gene, 239: 237–242.
Jessop, BM (1976). Distribution and timing of tag recoveries from native and nonnative Atlantic salmon (Salmo salar) released into Big Salmon R829–833ew Brunswick. J Fish Res Bd Canada, 33: 829–833.
King, TL, Spidle, AP, Eackles, MS, Lubinski, BA, Schill, WB (2000). Mitochondrial DNA diversity in North American and European Atlantic salmon with emphasis on the Downeast rivers of Maine. J Fish Biol, 57: 614–630.
Knox, D, Lehmann, K, Reddin, DG, Verspoor, E (2002). Genotyping of archival Atlantic salmon scales from northern Quebec and West Greenland using novel PCR primers for degraded mtDNA. J Fish Biol, 60: 266–270.
Koljonen, M -L, Jansson, H, Paaver, T, Vasin, O, Koskiniemi, J (1999). Phylogeographic lineages and differentiation pattern of Atlantic salmon (Salmo salar) in the Baltic Sea with management implications. Can J Fish Aquat Sci, 56: 1766–1780.
Kumar, S, Tamura, K, Jakobsen, IB, Nei, M (2001). MEGA2: Molecular Evolutionary Genetics Analysis software. Bioinformatics Applications Notes, 17: 1244–1245.
Maccrimmon, HR, Gots, BL World distribution of Atlantic salmon, Salmo salar. J Fish Res Bd Canada, 36: 422–457.
McConnell, SKJ, Ruzzante, DE, O’reilly, PT, Hamilton, L, Wright, JM (1997). Microsatellite loci reveal highly significant genetic differentiation among Atlantic salmon (Salmo salar L.) stocks from the east coast of Canada. Mol Ecol, 6: 1075–1089.
McElroy, D, Moran, P, Berminham, E, Kornfield, I (1991). REAP: The restriction enzyme analysis package, Ver 4.0. http://bioweb.wku.edu/faculty/mcelroy/.
Mills, D (1989). Ecology and Management of Atlantic salmon. Chapman and Hall: London.
Nielsen, J (1961). Contributions to the biology of the salmonidae in Greenland I-IV. Meddelelser om Grønland, 159, Nr 8.
Nilsson, J, Gross, R, Asplund, T, Dove, O, Jansson, H, Kellonieni, J et al. (2001). Matrilineal phylogeography of Atlantic salmon (Salmo salar L.) in Europe and post-glacial colonizat Baltic Sea area. Mol Ecol, 10: 89–102.
Orr, MR, Smith, TB (1998). Ecology and speciation. TREE, 13: 502–506.
Pielou, EC (1991). After the Ice Age: the return of life to glaciated North America. University of Chicago Press: Chicago.
Posada, D, Crandall, KA, Templeton, AR (2000). GeoDis: A program for the Cladistic Nested Analysis of the Geographical Distribution of Genetic Haplotypes. Mol Ecol, 9: 487–488.
Shaw, J, Gareau, P, Courtney, RC (2002). Palaeogeography of Atlantic Canada 13–0 kyr. Quaternary Sci Rev, 21: 1861–1878.
Stabell, OB (1984). Homing and olfaction in salmonids: a critical review with special reference to the Atlantic salmon. Biol Rev, 59: 333–388.
Ståhl, G (1987). Genetic population structure of Atlantic salmon. In: Ryman N, Utter F (eds) Population Genetics and Fishery Management, University of Washington Press: Seattle, pp 121–140.
Templeton, AR (1998). Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history. Molecular Ecology, 7: 381–397.
Templeton, AR, Sing, CF (1993). A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics, 134: 659–669.
Templeton, AR, Crandall, KA, Sing, CF (1992). A cladistic of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. III. Cladogram estimation. Genetics, 132: 619–633.
Templeton, AR, Routman, E, Phillips, CA (1995). Separating population structure from population history767–782 distic analysis of the geographical distribution of mitochondrial DNA haplotypes in the Tiger salamander, Ambystoma tigrinum. Genetics, 140: 767–782.
Tessier, N, Bernatchez, L, Wright, JM (1997). Population structure and i735–750f supportive breeding inferred from mitochondrial and microsatellite DNA analyses in land-locked Atlantic salmon Salmo salar L. Mol Ecol, 6: 735–750.
Verspoor, E (1986). Spatial correlations of transferrin allele frequencies in Atlantic salmon populations from North America. Can J Fish Aquat Sci, 43: 1074–1078.
Verspoor, E (1994). The evolution of genetic divergence at protein coding loci among anadromous and nonanadromous populations of Atlantic salmon Salmo salar. In: Beaumont AR Genetics on of Aquatic Organisms. Chapman-Hall: London pp 52–67.
Verspoor, E (1997). Genetic diversity among Atlantic salmon (Salmo salar L.) populations. ICES J Mar Sci, 54: 965–973.
Verspoor, E, Cole, LC (1989). Genetically distinct sympatric populations of resident and anadromous Atlantic salmon Salmo salar. Can J Zool, 67: 1453–1461.
Verspoor, E, McCarthy, E, Knox, D, Bourke, E, Cross, TF (1999). The Phylogeography of European Atlantic salmon (Salmo salar L.) based on RFLP analysis of the ND1/16sRNA region of the mtDNA. Biol J Linn Soc, 68: 129–146.
Wilder, DG (1947). A comparative study of the Atlantic salmon Salmo salar (Linnaeus) and the lake salmon, Salmo salar sebago (Girard). Can J Fish Res Section D, 25: 175–189.
Youngson, AF, Jordan, WC, Hay, DW (1994). Homing of Atlantic salmon (Salmo salar L.) to a tributary stream in a major river catchment. Aquaculture, 121: 259–267.
Acknowledgements
The assistance of Dave Longard and others in collection of the samples in the field is gratefully acknowledged. Samples of hatchery fish were kindly provided by Bev Davisson and Trevor Goff while Tim King generously made available the sample collected from the Narraguagus River. The work would not have been possible without the support of John Ritter. Patrick O’Reilly and Stuart Piertney kindly reviewed an early draft of the manuscript. John Brookfield’s comments on the manuscript helped to clarify many aspects of the discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Verspoor, E., O'sullivan, M., Arnold, A. et al. Restricted matrilineal gene flow and regional differentiation among Atlantic salmon (Salmo salar L.) populations within the Bay of Fundy, eastern Canada. Heredity 89, 465–472 (2002). https://doi.org/10.1038/sj.hdy.6800166
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6800166
Keywords
This article is cited by
-
Comparative estuarine and marine migration ecology of Atlantic salmon and steelhead: blue highways and open plains
Reviews in Fish Biology and Fisheries (2014)
-
Use of microsatellite data and pedigree information in the genetic management of two long-term salmon conservation programs
Reviews in Fish Biology and Fisheries (2014)
-
Adaptive strategies and life history characteristics in a warming climate: Salmon in the Arctic?
Environmental Biology of Fishes (2013)
-
Detection and mapping of mtDNA SNPs in Atlantic salmon using high throughput DNA sequencing
BMC Genomics (2011)
-
Genetic stock identification of Atlantic salmon (Salmo salar) populations in the southern part of the European range
BMC Genetics (2010)
