
Abstract
The geographic distribution of haplotype diversity in the rodent Calomys musculinus, sampled from 16 wild populations of Argentina, was analysed on two geographical scales. The species is the natural reservoir of the Junin virus, the etiological agent of the Argentine haemorrhagic fever (AHF). In all, 24 composite haplotypes were recognised in the mtDNA D-loop region. Haplotypes 1 and 2, internal in the network, were the most frequent and were present in almost all populations. The absence of large genetic gaps between widely distributed haplotypes, the existence of exclusive haplotypes in more than 50% of the sampled populations and the absence of isolation by distance at a macrogeographical scale are in support of the hypothesis of a recent range expansion of the populations of the Humid Pampa, with low to moderate current gene flow. The dispersal of this opportunistic species would have been favoured by the explosive increments in density after agriculture was introduced. When only nearby populations within the endemic area of AHF were considered, a pattern of isolation by distance was detected. At present, genetic drift appears to be the main force acting to randomly differentiate C. musculinus populations, which would also lead to random differentiation of Junin virus strains and a reduction in the virulence of the pathogen in ‘historic’ AHF areas. The knowledge of migration patterns of the reservoir populations facilitates reliable prediction of the potential spread of the human disease.
Similar content being viewed by others
Introduction
The analysis of the genetic similarity of different populations has traditionally enabled the estimation of gene flow, one of the determinants of population genetic structure. In recent years, a different approach has been extensively used to evaluate the incidence of life-history traits and ongoing genetic exchange in observed patterns of genetic diversity. Phylogeographic methods, which combine intraspecific gene phylogeny with spatial distribution of variants, have had a major impact on research addressing the evaluation of processes shaping the genetic structure of populations (Schaal et al, 1998; Avise, 2000).
Besides its evolutionary interest, the phylogeographic approach is useful when applied to the study of genetic structure of rodent populations that act as hosts of disease affecting humans. The murid Calomys musculinus (Thomas, 1913) is the reservoir of the Junin virus, the etiological agent of Argentine haemorrhagic fever (AHF) (Parodi et al, 1958; Calderón, 2004). Since this endemic disease was detected in 1955 in the extensive agroecosystem of the central-eastern plains of Argentina, its range has been expanding steadily and the endemic area now comprises more than 150 000 km2 (Enría et al, 1998). The incidence of the disease varies over time: in general, it is highest during a period of 5–10 years in newly affected areas and then gradually declines and, in some cases, disappears. The prevalence of infection in reservoir populations may be very high in some localities and very low or absent at nearby sites, suggesting that the virus periodically becomes extinct in local reservoir populations but is later reintroduced from neighbouring ones (Mills and Childs, 1998).
C. musculinus is an opportunistic species; it occupies a variety of habitats, including crop fields, where its population may be very large (Busch et al, 2000). Virus transmission occurs when farmers working in the fields inhale aerosols of virus from urine, faeces or saliva of infected rodents, which transmit the infection by contact with other animals, mainly in the course of fights between adult males during reproductive periods (Sabattini et al, 1977).
Since the possibility of virus transmission between individuals of the reservoir species is related to the degree and direction of gene flow among populations, knowledge of the colonisation patterns of C. musculinus could contribute to our understanding of the epidemiology of AHF.
Chiappero et al (2002) and Chiappero and Gardenal (2003) studied the genetic structure of C. musculinus populations from the central-eastern plains of Argentina using allozymes and RAPDs. On the basis of geographic distribution patterns of genetic polymorphisms, the authors proposed that differentiation by genetic drift proceeds faster than homogenisation by gene flow, in spite of the high Nm values obtained. In a previous paper, we described the utility of the D-loop region of mitochondrial DNA as a genetic marker at the population level in this species. On the basis of restriction fragment length polymorphism (RFLP-PCR), 20 different composite haplotypes were detected, only two of which were very frequent and common to all the five populations analysed (González Ittig and Gardenal, 2002).
In the present study, we analysed 16 populations: nine were from the endemic area of AHF, two were from the marginal zone and the other five were from locations outside that area. In order to evaluate the relative influence of current gene flow on the population structure of C. musculinus, we studied the degree of genetic divergence between haplotypes in relation to their distribution in Argentina on two different geographic scales. Our aim was to gain insight into the processes determining the geographic expansion of the endemic area and the changing incidence of AHF.
Materials and methods
Sample collection
A total of 231 specimens of C. musculinus were lived-trapped in 16 localities of Argentina (Figure 1), distributed as follows: (A) populations from the endemic zone of AHF: Pergamino (n=15), Rojas (n=7), Zárate (n=19), San Nicolás (n=13), San Pedro (n=17), Melo (n=18), Uranga (n=13), Alcorta (n=19) and JB Molina (n=13). (B) Populations from the marginal zone: Oliveros (n=6) and Maciel (n=8). These are located in the central-eastern plains of Argentina in the phytogeographic region known as ‘Humid Pampa’, a temperate grassland with annual rainfall of more than 1200 mm that has been intensively disturbed by agriculture and other human activities since the 1880s. (C) Populations from outside the AHF area: the fields of the Agronomy Faculty, National University of Cordoba (n=10), Laguna Larga (n=18), Parque Luro Wildlife Reservation (n=20), which are situated in the phytogeographic region called ‘Espinal’, a plain at low elevation (600–900 m) characterised by an annual rainfall of less than 600 mm, with vegetation dominated by ‘algarrobo’ trees (Prosopis alba, P. nigra). Donovan (n=16) and Molinari (n=19) belong to the ‘Chaco Serrano’, which is a xerophytic woodland with an altitude between 600 m in the valleys and 1800 m in the mountains (annual rainfall of 500–800 mm). The main tree species are Schinopsis haenkeana (horco-quebracho), Lithrea ternifolia (molle) and Fagara coco. In recent years, the latter two regions have suffered marked alterations as a result of cattle farming and agriculture.

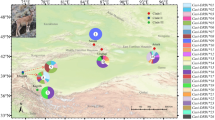
Geographic distribution of C. musculinus populations from different phytogeographic regions (Pampa, Espinal, Chaco Serrano). Circles represent the relative frequencies of haplotypes 1, 2 and a pool of the other haplotypes present at each locality. 1: Donovan, 2: Molinari, 3: Laguna Larga, 4: Melo, 5: Maciel, 6: Oliveros, 7: Uranga, 8: Alcorta, 9: J. B. Molina, 10: San Nicolás, 11: Pergamino, 12: San Pedro, 13: Rojas, 14: Zárate, 15: Parque Luro, 16: Agronomy Faculty fields. The grey-shaded area in the Pampa indicates the endemic zone of AHF.
Specimens from the Humid Pampa area and from Parque Luro are preserved in the Instituto Nacional de Enfermedades Virales Humanas (INEVH), Pergamino, Buenos Aires Province. Animals from Donovan are deposited in the Department of Animal Ecophysiology, National University of San Luis, Argentina, and those from Molinari, Laguna Larga and the Agronomy Faculty fields are preserved in the Museum of Zoology, National University of Cordoba.
DNA extraction
DNA from individual samples of liver or kidney was obtained by the CTAB protocol described in Milligan (1992). Tissues were handled in a vertical laminar flow cabinet, taking the appropriate precautions required to deal with potentially infected materials.
Mitochondrial DNA amplification
The noncoding control region, or ‘D-loop’, was amplified using primers with the following sequences: 5′-AAGGCTAGGACCAAACCT-3′ and 5′-TGAATTGGAGGACAACCAGT-3′. The amplification conditions in a 50 μl volume were those described by González Ittig and Gardenal (2002): 240 μM each of dATP, dGTP, dCTP, dTTP; 200 nM of each primer, 1 × reaction buffer, 2.5 mM MgCl2, 1.0 U of Taq polymerase (Amersham Biosciences) and between 10 and 25 ng of total DNA. The reaction started with denaturation at 94°C for 3 min, followed by 35 cycles of denaturation for 30 s at 94°C, annealing for 90 s at 55°C and extension for 90 s at 72°C. Finally, there was a hold period of 5 min at 72°C.
Restriction enzyme digestion
Between 3 and 5 μg of each PCR product in a final volume of 25 μl were digested separately with the endonucleases restriction selected previously (González Ittig and Gardenal, 2002). Restriction fragments were separated by horizontal 2% agarose gel electrophoresis. Gels were stained with ethidium bromide and photographed under UV light. Fragment sizes were estimated by comparison with a 100 bp ladder (Gibco BRL) and a 1 kb ladder (Promega).
Restriction site inference
Restriction sites were inferred from patterns of fragment length polymorphisms obtained with each enzyme. A few fragments smaller than 100 bp could not be resolved, but were inferred from pattern analysis. In DNA samples, the sum of the fragments was greater than the total size of the amplified D-loop region. In those cases, the PCR product from single individuals was cloned in a pGEM-T Easy Vector (Promega, Madison). Standard plasmid miniprep procedures allowed recovery of plasmid DNA; presence of the insert was corroborated by specific PCR amplification using the primers described above. These PCR products were digested and restriction patterns for each clone obtained.
Statistical analysis
Patterns for each enzyme were named with capital letters and composite haplotypes were designated by numbers. Haplotypes resulting from cloning experiments are indicated by an asterisk (Tables 1 and 2).
Using the REAP software package (McElroy et al, 1992), a presence–absence matrix for restriction sites for each composite haplotype was derived with the GENERATE program and genetic distances (Nei and Miller, 1990) between the haplotypes were calculated with the D program.
A network was constructed with the program MINSPNET (Excoffier et al, 1992). A hierarchical analysis of population structure was performed using AMOVA in Arlequin Version 1 (Schneider et al, 1997). Besides an analysis of all collection sites, populations were grouped according to the phytogeographic region and whether inside or outside the AHF endemic zone.
The pattern of isolation by distance was investigated by the correlation between pairwise comparisons of the genetic distance and the natural logarithm of the geographic distances, using the Mantel test in TFPGA Version 1.3 (Miller, 1998): (a) considering all the populations (macrogeographic scale); (b) considering only the eight relatively close populations (30–200 km apart) within the endemic AHF area (populations 7–14 in Figure 1).
Results
The amplification product was 1300 bp long in all the 231 individuals analysed. Nine of the 16 restriction enzymes used produced monomorphic patterns (AccI, ApaI, MspI, NciI, NdeI, Sau3AI, Sau96I, TaqαI, AluI), whereas the following enzymes revealed fragment length polymorphisms: RsaI, AciI, MseI, HaeIII, AseI, Tsp509I and NlaIII. The enzyme RsaI yielded seven different patterns; AseI, NlaIII, HaeIII and MseI revealed three; AciI and Tsp509I showed two.
In our previous paper on the haplotype diversity of the D-loop region in C. musculinus (González Ittig and Gardenal, 2002), we included patterns that were the result of the existence of at least two different mitochondrial clones in the same individual (eg pattern B for AluI, pattern D for RsaI and pattern D for MseI). The samples analysed in the present work allowed new cases of heteroplasmy to be identified when the sum of the fragment sizes was greater than the total size of the amplified region. Each identified clone was then treated as a separate haplotype. As a consequence, there are minor differences in haplotype nomenclature compared with the previous report.
Table 1 summarises patterns obtained with restriction enzymes revealing polymorphisms; 24 composite haplotypes were determined among the 247 clones derived from the 231 specimens examined. The number of mitochondrial clones with a particular haplotype from each locality is presented in Table 2. Haplotypes 1 and 2 were the most abundant (with overall frequencies of 40 and 34%, respectively) and were found in the majority of the populations, except that of the Agronomy Faculty, from which haplotype 2 was absent (Figure 1). Haplotype 13, with a frequency of 9%, was present in eight populations and was more abundant than haplotypes 1 or 2 in Zárate, Molinari, Donovan and Parque Luro. Haplotypes 9*, 12*, 14, 15, 18* and 26, present at low frequencies, occurred in two or more distant populations. Nine out of 16 populations featured unique haplotypes (Table 2).
The network of the relationships and relative abundance of the 24 haplotypes is shown in Figure 2. Haplotypes 1 and 2 occupied a central position, forming a ‘two-star phylogeny’ (Avise, 2000). Most haplotypes were related by only one restriction site. Haplotypes 12*, 18*, 19* and 20* (detected only in the heteroplasmic state) clustered together, with one or two restriction sites in common (Figure 2).

Minimum spanning network showing the relationships and relative abundance of the 24 haplotypes (indicated by a number) detected in 16 populations of C. musculinus. Each bar through the solid line represents one restriction site mutation between haplotypes. Dashed lines indicate alternative relationships.
When haplotype frequencies were compared among all the populations, significant differences were found (Fst=0.1, P<0.01); the Mantel test statistic revealed no significant correlation between genetic and geographic distances.
Populations were grouped according to their phytogeographic origin (Humid Pampa, Chaco Serrano or Espinal) and a hierarchical analysis was performed. Of the total variation, 2.95% was attributable to between-region variation (not significant), while 89% was contained within local populations (Table 3).
Populations belonging to the endemic area of AHF did not have haplotype frequencies significantly different from populations of the marginal AHF zone. There was a positive correlation between the natural logarithm of the genetic and geographic distances when only the eight close-together populations of the endemic area (populations 7–14, Figure 1) were considered (r=0.41; P<0.05). There was also significant between-population variation in haplotype frequencies (Fst=0.05; P<0.05).
Discussion
In this analysis of the distribution of variation in the mitochondrial control region of C. musculinus over most of its geographical range in Argentina, a total of 24 haplotypes were found in 231 specimens. Cloning experiments allowed us to separate different components from some individual PCR products. All of them were computed as single haplotypes, assuming them to be the result of heteroplasmy; this situation was found in 7% of the individuals analysed. It has also been reported in high proportions in other mammals like rabbits (Casane et al, 1994) and bats (with an incidence of 15–63%; Wilkinson et al, 1997). Zullo et al (1991) demonstrated that 0.5 kb segments showing 80–88% homology with a section of the D-loop region were integrated in the rat nuclear genome as pseudogenes. The transposition of mtDNA sequences to the nuclear DNA could lead to co-amplification of those fragments, when universal primers for PCR are applied to total DNA extracts. We sequenced the D-loop region from clones with haplotypes 1 and 2 and compared them with those present only in the heteroplasmic state; they showed very slight nucleotide divergence (from 0.01 to 0.02) (unpublished results), suggesting that they do not represent pseudogenes, but originate from mitochondrial DNA mutations within an isolated female lineage or within the somatic cells of the individual.
The network connecting the different haplotypes has no large genetic gaps (maximum of two mutational steps) and two common widespread lineages, haplotypes 1 and 2, at frequencies of 40 and 34%, respectively; they occupy a central position in the network and very probably represent ancestral-like forms. Nine of the 16 populations have exclusive haplotypes; most of them are rare and can be considered derived forms (Figure 2). This phylogeographic outcome is compatible with the pattern proposed as category V by Avise (2000) for populations that have increased rapidly in size as the species expanded its range in recent evolutionary time from a few founders, and with contemporary gene flow among them being low to moderate.
The genus Calomys would have originated in the high Andean plains of the Puna by the late Miocene, and then speciated during its dispersal to the eastern lowlands (Reig, 1984). The first fossil record of Calomys species in the Humid Pampa is from the Middle Pleistocene (Tonni et al, 1993). From this period until the upper Holocene, the species was recorded at a very low frequency in the area (Pardiñas et al, 2002). Bilenca and Kravetz (1995) suggested that, since the introduction of agriculture in the Humid Pampa (at the end of the 19th century), corridors of habitats favourable to the species movements would have been available. In recent decades, the Espinal and Chaco Serrano regions have also been disturbed by cultivation. Given that C. musculinus is a better coloniser than the other co-inhabiting rodent species (genera Akodon, Mus and Necromys), its populations can attain very high densities in disturbed habitats like crop fields (Busch and Kravetz, 1992). The absence of a well-defined geographic structure in the rodent populations, as revealed in this study, could be explained by population expansions after explosive increments in density caused by the introduction of agriculture. Once established in an area, high adult mortality occurs during winter and the juvenile survivors initiate a new population cycle in the following spring (de Villafañe and Bonaventura, 1987). Population bottlenecks and low levels of gene flow would favour genetic differentiation among demes by genetic drift. The latter would be the main force acting to differentiate even close populations, as suggested by the fact that some samples (eg, San Pedro, Uranga, San Nicolás and Zárate) present ‘private’ haplotypes that were not detected in any nearby population.
The recent geographic expansion of the endemic area of AHF has been slower than in the period immediately after the discovery of the disease, suggesting some self-limiting mechanism of expansion. This observation is also consistent with low to moderate levels of contemporary gene flow in C. musculinus.
García et al (2000) studied genetic diversity in strains of Junin Virus from Central Argentina, derived from human and rodent isolates. The strains were grouped in three distinct clades: the first clade included 33 strains from the centre of the endemic area of AHF; the second clade contained four strains from Zárate (the north-eastern edge of the endemic area); the third group consisted of four strains from Melo and two nearby localities (south-west of the endemic area). We found no similar geographic structure of the genetic diversity, since haplotypes from Zárate and Melo exhibited no particular clustering.
As mentioned before, the incidence of the disease can be high for 5–10 years and then it slowly declines or disappears (‘historic’ areas) (Enría et al, 1998). A close relationship between population ‘flushes’ of C. musculinus and the increment of AHF cases has been observed; after population ‘crashes’ in winter or during harvest, a bottleneck in the viral populations would also occur (Mills et al, 1992). As our results indicate, genetic drift in these periods would randomly differentiate the geographic distribution of C. musculinus haplotypes. The same process would also promote random differentiation of Junin virus strains and a decrease in horizontal transmission among rodents, which would contribute to the reduction of the virulence of the pathogen in ‘historic’ areas (Calderón, 2004).
Using allozymes as genetic markers, Chiappero et al (2002) estimated that the effective number of migrants (Nm) was between 2.9 and infinity for populations of C. musculinus from the Humid Pampa, with significant overall differentiation populations (θ=0.020; P<0.01). The authors did not find correlation between genetic and geographic distances, suggesting that the species has expanded its range relatively recently. In the present study, when the eight nearby populations from the endemic area of AHF were considered, a low but significant r value was obtained. Hutchison and Templeton (1999) proposed four models to summarise hypothetical relationships between geographic and genetic distances, taking into account the scattering of points in a pairwise representation. Model IV predicts a stronger relative influence of genetic drift than gene flow at greater distances of geographic separation (as indicated by highly scattered points); gene flow would be more effective than drift between neighbouring populations (little scatter of points over shorter distances). Our results fit this model (Figure 3).

Scatterplot of pairwise genetic distances vs. natural logarithm of geographic distances (in km) between close populations of Calomys musculinus from the endemic area of AHF.
The limits of the endemic area of AHF are defined by the occurrence of the disease in humans, but infected rodents have been detected beyond those limits (Enría et al, 1998). Given the predominance of current genetic drift over gene flow in reservoir populations, as indicated by the results presented here and those of previous papers (Chiappero et al, 2002; Chiappero and Gardenal, 2003), a steady and uniform expansion of the geographic range of infection by Junin virus would be unexpected. Our results support the hypothesis that focal emergence or re-emergence of the disease is likely to occur in localities where the pathogen has been detected and where habitat modifications by human activities, combined with favourable weather conditions, determine sudden increases in C. musculinus population density (Mills et al, 1992; Calderón, 2004). These would increase the probability of horizontal transmission of Junin virus.
According to de Villafañe and Bonaventura (1987), C. musculinus individuals rarely move more than 200 m, which suggests a low dispersal capacity in the species. A microgeographic-scale study using microsatellites is in progress in our laboratory with the aim of increasing our understanding of patterns of migration of the rodent, since this is one of the factors that must be considered in order to explain the expansion and changing incidence of the AHF.
References
Avise JC (2000). Phylogeography. The History and Formation of Species, Harvard University Press: USA.
Bilenca DN, Kravetz FO (1995). Patrones de abundancia relativa en ensambles de pequeños roedores de la Region Pampeana. Ecol Austral 5: 21–30.
Busch M, Kravetz FO (1992). Competitive interactions among rodents (Akodon azarae, Calomys laucha, Calomys musculinus and Oligoryzomys flavescens) in a two-habitat system. II. Effect of species removal. Mammalia 56: 545–554.
Busch M, Miño MH, Dadon JR, Hodara K (2000). Habitat selection by Calomys musculinus (Muridae, Sigmodontinae) in crop areas of the pampean region, Argentina. Ecol Austral 10: 15–26.
Calderón DE (2004). Desarrollo de indicadores de riesgo de contraer la FHA por medio del estudio de los roedores que actúan como reservorio de los arenavirus en Argentina. Doctoral Thesis, Universidad Nacional del Litoral.
Casane D, Dennebouy N, de Rochambeau H, Mounolou JC, Monnerot M (1994). Genetic analysis of systematic mitochondrial heteroplasmy in rabbits. Genetics 138: 471–480.
Chiappero MB, Sabattini MS, Blanco A, Calderón GE, Gardenal CN (2002). Gene flow among Calomys musculinus (Rodentia, Muridae) populations in Argentina. Genetica 114: 63–72.
Chiappero MB, Gardenal CN (2003). Restricted gene flow in Calomys musculinus (Rodentia, Muridae), the natural reservoir of Junin Virus. J Heredity 94: 490–495.
de Villafañe G, Bonaventura SM (1987). Ecological studies in crop fields of the endemic area of Argentine hemorrhagic fever. Calomys musculinus movements in relation to habitat and abundance. Mammalia 51: 233–248.
Enría DA, Briggiler AM, Feuillade MR (1998). An overview of the epidemiological, ecological and preventive hallmarks of Argentine haemorrhagic fever (Junin virus). Bull Inst Pasteur 96: 103–114.
Excoffier L, Smouse PE, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
García JB, Morzunov SP, Levis S, Rowe J, Calderon G, Enría D et al (2000). Genetic diversity of the Junin virus in Argentina: geographic and temporal patterns. Virology 272: 127–136.
González Ittig RE, Gardenal CN (2002). Haplotype diversity of the mitochondrial DNA D-loop region in Calomys musculinus (Rodentia, Muridae) detected by PCR-RFLP. Biochem Genet 40: 293–302.
Hutchison DW, Templeton AR (1999). Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53: 1898–1914.
McElroy D, Moran P, Bermingham E, Kornfield I (1992). REAP: an integrated environment for the manipulation and phylogenetic analysis of restriction data. J Heredity 83: 157–158.
Miller M (1998). Tools for population genetic analyzes (TFPGA), Version 1.3. A window program for the analysis of allozyme and molecular population genetic data. Available at: URL: http://bioweb.usu.edu/mpmbio/tfpga.asp.
Milligan BG (1992). Plant DNA isolation. In: Hoelzel AR (ed) Molecular Genetic Analysis of Populations: A Practical Approach, IRL Press: Oxford. pp 59–88.
Mills JN, Childs JE (1998). Ecologic studies of rodent reservoirs: their relevance for human health. Emerging Infect Dis 4: 529–537.
Mills JN, Ellis BA, McKee KT, Calderón GE, Maiztegui JI, Nelson GO et al (1992). A longitudinal study of Junin virus activity in the rodent reservoir of Argentine hemorrhagic fever. Am J Trop Med Hygiene 47: 749–763.
Nei M, Miller JC (1990). A simple method for estimating average number of nucleotide substitutions within and between populations from restriction data. Genetics 125: 873–879.
Pardiñas UFJ, D'Elía G, Ortiz PE (2002). Sigmodontinos fósiles (Rodentia, Muroidea, Sigmodontinae) de América del Sur: estado actual de su conocimiento y prospectiva. Mastozool Neotropical 9: 209–252.
Parodi AS, Geenway DY, Rugiero HR, Rivero E, Frigerio MJ, Mettler NE (1958). Sobre la etiologia del brote epidémico en Junin. Dia Medico 30: 2300–2302.
Reig OA (1984). Ditribuiçáo geográfica e historia evolutiva dos roedores muroideos sulamericanos (Cricetidae: Sigmodontinae). Rev Brasil Genet 7: 333–365.
Sabattini MS, Gonzalez de Ríos L, Díaz G, Vega R (1977). Infección natural y experimental de roedores con Virus Junin. Medicina (Buenos Aires) 37 (Suppl. 3): 149–159.
Schaal BA, Hayworth DA, Olsen KM, Rauscher JT, Smith WA (1998). Pylogeographic studies in plants: problems and prospects. Mol Ecol 7: 465–474.
Schneider S, Kueffer JM, Roessli D, Excoffier L (1997). Arlequin ver 1.1. A software for population genetic data analysis Genetics and Biometry Laboratory, University of Geneva, Switzerland.
Tonni EP, Verzi DH, Bargo MS, Pardiñas UFJ (1993). Micromammals in owl pellets from the lower-middle Pleistocene in Buenos Aires province, Argentina. Ameghiniana 30: 342.
Wilkinson GS, Mayer F, Kerth G, Petri B (1997). Evolution of repeated sequence arrays in the D-loop region of Bat mitochondrial DNA. Genetics 146: 1035–1048.
Zullo S, Sieu LC, Slightom JL, Hadler HI, Eisenstadt JM (1991). Mitochondrial D-loop sequences are integrated in the rat nuclear genome. J Mol Biol 221: 1223–1235.
Acknowledgements
We thank Drs MS Sabattini and G Calderón from the INEVH (Pergamino, Argentina) for providing most of the rodent samples analysed here and Dr Caviedes Vidal from the National University of San Luis, Argentina, for providing animals from Donovan. We are also grateful to Dr A Blanco for critical revision of the manuscript and to Dr AR Templeton for advice on the analysis of some of the data. This work was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina, the Agencia Córdoba Ciencia and the Secretaría de Ciencia y Tecnología (SECYT), Universidad Nacional de Córdoba, Argentina. REGI is a Fellow and CNG is a Career Investigator of CONICET.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
González Ittig, R., Gardenal, C. Recent range expansion and low levels of contemporary gene flow in Calomys musculinus: its relationship with the emergence and spread of Argentine haemorrhagic fever. Heredity 93, 535–541 (2004). https://doi.org/10.1038/sj.hdy.6800546
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6800546
