
Abstract
We investigated 25 natural populations of Chamaecyparis obtusa using 51 cleaved amplified polymorphic sequence (CAPS) markers, which were developed using information on sequence-tagged sites (STS) in Cryptomeria japonica. Most CAPS markers have codominant expression patterns, and are suitable for population studies because of their robustness and convenience. We estimated various genetic diversity parameters, including average heterozygosity (He) and allelic richness and found that the more peripheral populations tended to have lower genetic diversity than central populations, in agreement with a previous theoretical study. The overall genetic differentiation between populations was low, but statistically significant (GST=0.039), and similar to the level reported in a previous allozyme study. We attempted to detect non-neutral loci associated with local adaptation to clarify the relationship between the fixation index (FST) and He values for each locus and found seven candidates non-neutral loci. Phylogenetic tree analysis of the populations and Bayesian clustering analysis revealed a pattern of gradually increasing isolation of populations with increasing geographical distance. Three populations had a high degree of linkage disequilibrium, which we attribute to severe bottlenecks due to human disturbance or competition with other species during their migration from refugia after the most recent glaciation. We concluded that the small populations in western Japan and in Kanto district are more important, from a conservation perspective, than the populations in central Japan, due to their genetic divergence, relatively small sizes and restricted areas.
Similar content being viewed by others

Introduction
Natural forests have become fragmented and patchily distributed owing to human activities such as logging, clearance for agricultural purposes and intensive, monocultural forestry. Such disturbance of once-continuous tree populations could potentially disrupt natural ecological and evolutionary processes and adversely modify their genetic structure (Hamrick, 2004). Species that are widely used in forestry may also be affected by contamination from artificial forests of conspecific species, and consequent modification of their genetic structure. Thus, to facilitate genetic conservation efforts we should ideally clarify the genetic structure of these species before such effects occur.
The genetic structure of forest tree species sometimes shows clear geographical clines in genetic diversity that can provide insights into the population history of the species, including the establishment and migration of their populations. This has been demonstrated for two European species: Fagus sylvatica (Comps et al., 2001) and Picea abies (Lagercrantz and Ryman, 1990). In addition, Tomaru et al. (1997) demonstrated a clear genetic cline with respect to latitude in Fagus crenata populations in Japan, and suggested that populations of this species were distributed in western Japan during the last glacial period then rapidly expanded northward during the postglacial period. Population genetics studies of Japanese Abies species (Tsumura and Suyama, 1998) and Pinus pumila (Tani et al., 1996) have found similar trends to that shown by F. crenata (for a review, see Tsumura, 2006). Most of these results have been supported by evidence from fossil pollen (Tsukada, 1983, 1988). However, identification of pollen in some species is very difficult because of its morphological similarity to that of other species. For instance, Chamaecyparis obtusa pollen cannot be easily identified because pollen from members of three families (Cupressaceae (including C. obtusa), Taxaceae and Cephalotaxaceae; C-T-C) are very similar and difficult to distinguish (H. Takahara, personal communication). Thus, their pollen is collectively identified as C-T-C type. Consequently, the population history of C. obtusa, and its refugia during the last glacial period, cannot (as yet at least) be inferred from fossil pollen records. Thus, other methods involving population genetic analyses have to be used in attempts to clarify the population history of this species.
Elucidating the genetic structure of such natural forest populations is important not only for conservation purposes, but also to optimize their use as breeding materials. Elucidating how current natural forests of certain species have established since the last glacial period is especially important for the conservation of local populations. The identification of genes related to local adaptation and differentiation is also essential for the conservation of local populations. However, such genes have only been identified in model plants to date (for a review, see Shimizu and Purugganan, 2005), although genomic regions associated with speciation and population differentiation have been reported in several forest tree species, including Quercus and Picea abies (Scotti-Saintagne et al., 2004; Acheré et al., 2005). Thus, there is an urgent need to extend the identification of such genes to the genomes of other species to facilitate conservation efforts.
C. obtusa, hinoki, one of the most important Japanese conifers, belongs to the Cupressaceae family. Current natural forests of this species are scattered in isolated locations ranging from Fukushima Prefecture (37°10′N) in north-central Japan to Yakushima Island (30°15′N) in southern Japan, and large natural forests have only been retained in the Kiso area (35°34′–36°35′N) of central Japan. The wood of this species has been used for house construction since at least the Nara era (AD 710; Suzuki, 2002), and possibly longer, because of its high quality for use as pillars and beams. As the demand for timber production increased, the natural forests were rapidly depleted, particularly after World War II. As a result, many natural forests have decreased greatly in extent and, even in the Kiso area, many large trees have been selectively logged from the natural forests.
Previous population studies of this species using allozyme markers have found low degrees of genetic differentiation between populations (GST=0.045, Uchida et al., 1997) and evidence of a genetic cline in allele frequency at a few loci (Shiraishi et al., 1987). The genetic relationship between populations mostly followed their population locations based on genetic distances (Uchida et al., 1997). The cited authors compared the genetic diversity between natural populations and plus-tree populations in which superior individuals (based on growth and other traits) were selected by Japan's Forestry Agency to serve as sources for seed orchards and breeding materials. The level of genetic diversity was very similar between these two groups of trees, but the plus-tree groups were most genetically similar to the natural populations found in central Japan. However, Uchida et al. (1997) investigated only 11 populations using a limited number of loci. To elucidate the genetic structure of this species more precisely, a more comprehensive study is required, in which many more populations throughout the natural distribution of the species are investigated using a sufficient number of loci.
We developed and evaluated STS primers based on EST information derived from sugi (Cryptomeria japonica) for use in hinoki (C. obtusa) and found that 30% of the examined C. japonica STS primers were directly transferable to C. obtusa (Matsumoto and Tsumura, 2004). We have applied the previously identified CAPS markers in a population study of C. obtusa to detect non-neutral loci and the genetic structure of C. obtusa. In this paper, we focus on the genetic diversity and structure of the surviving C. obtusa natural populations. We discuss the putative establishment of the current natural forests of C. obtusa and the importance of natural forests of this species.
Materials and methods
Plant materials
We collected fresh leaf tissues from 481 individuals representing 25 natural populations of C. obtusa. All trees were sampled in national or private forests that represented in situ gene-conservation forests (Figure 1, Table 1). The locations of the sampled populations covered most of the natural distribution of C. obtusa. We collected leaf tissues from individual trees with considerable space between sampled specimens to avoid sampling half-sib individuals from the same mother tree. We also paid attention to the age and size of trees, because large-scale planting of C. obtusa began after 1945 in Japan; we therefore sampled the oldest and largest trees to eliminate or at least minimize the influence of gene flow from artificially selected planted populations. Collected leaf tissues were stored at −30°C before DNA extraction.

Natural distribution of C. obtusa in Japan (shaded areas; from Hayashi, 1960) and the locations of the 25 natural populations surveyed in the present study. Population 1 is in the South Tohoku district, 2, 3 and 4 in the Kanto district; 5–13 in the Chubu district; 14–17 in the Kinki district; 18, 19 and 22 in the Chugoku district; 20 and 21 in the Shikoku district; and 23–25 in the Kyushu district.
Investigated loci
Total DNA was extracted from all samples using the slightly modified CTAB method of Tsumura et al. (1995) or a DNeasy plant mini-kit (Qiagen GmbH, Hilden, Germany). The crude DNA was purified using a High Pure PCR Template Preparation Kit (Roche Co., Basel, Switzerland) when the quality of extracted DNA was insufficient for PCR. We investigated 51 CAPS markers that had been developed for C. obtusa using STS primers of C. japonica (Matsumoto and Tsumura, 2004; Table 2). We performed PCR amplifications in 20 μl of reaction volumes containing 0.2 μ M of each primer, 0.2 mM of each dNTP, 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 1.5 or 2.0 mM MgCl2, 0.4 U of Taq polymerase, and 4 ng of template DNA using thermal cyclers (GeneAmp PCR Systems 9600 or 9700, PE Applied Biosystems Co., Foster City, CA, USA) with the following temperature program: 5 min at 94°C, 35–45 cycles of 1 min at 94°C, 1 min at 50–65°C and 1 min 30 s at 72°C, with a final extension step of 5 min at 72°C. The concentration of MgCl2, the annealing temperature, and the number of cycles were optimized for the amplification of specific STS fragments.
The PCR products were digested with restriction enzymes specific for each CAPS marker (Table 2) and their restriction patterns were then checked by electrophoresis in 2% agarose gels followed by ethidium bromide staining.
Sequences of CAPS loci in C. obtusa were compared to known amino-acid sequences in the NCBI NR database, using a threshold BLASTX expected value (E value) of <10−10 to infer the putative function of the CAPS markers we used (Matsumoto and Tsumura, 2004; Ujino-Ihara et al., 2005). We also searched for tentative unique genes with significant similarity to infer the copy number of each gene using non-redundant cDNA databases for C. obtusa, C. japonica, Pinus taeda, Arabidopsis thaliana and Oryza sativa by the method of Ujino-Ihara et al. (2005).
Genetic diversity and differentiation between and within populations
To estimate the level of genetic diversity in each population, we used the following measures calculated from the allele frequencies of all loci that we analyzed: the proportion of polymorphic loci (Pl) at the 95% probability level, the average number of alleles per locus (Na), unbiased heterozygosity (He; Nei, 1978), observed heterozygosity (Ho) and allelic richness (Rs; El Mousadik and Petit, 1996). We determined the fixation index to evaluate deviations from random mating, FIS (=1–[Ho/He]) for each polymorphic locus, and its average over all loci, to compare the observed genotype frequencies with the expected values assuming Hardy–Weinberg equilibrium (Wright, 1922; Nei and Chesser, 1983). We also analyzed deviations from expected values using Fisher's exact test. Total gene diversity (HT) represents the sum of the genetic variation within (HS) and among (DST) populations: HT=HS+DST. The coefficients of gene differentiation (GST) among populations were then calculated to determine how gene diversity was partitioned at each level using the equation GST=DST/HT (Nei, 1973). Coefficients of linkage disequilibrium were calculated for all pairs of loci using the method of Weir (1979, 1990) based on genotype data from each of the test populations. The variance of the estimates was also calculated, and differences from equilibrium were verified using the χ2 test (Weir, 1990). These analyses were done using GDA (Lewis and Zaykin, 2002) and FSTAT (Goudet, 2000) software.
Genetic structure
We calculated Nei's unbiased genetic distances for each population pair (Nei, 1978), constructed a dendrogram by means of the neighbor-joining method (Saitou and Nei, 1987) and performed bootstrap analysis using PHYLIP software (Felsenstein, 1995). To examine genetic differentiation between the central and marginal populations, we performed a hierarchical analysis of molecular variance (AMOVA; Excoffier et al., 1992) using Arlequin (Schneider et al., 1997), in which significance levels of the overall values were tested through the use of permutations. We also calculated genetic differentiation among populations by AMOVA.
To detect the population structure and infer the most appropriate number of populations (K) for interpreting the data without prior information on the number of locations at which the populations were sampled, we used the Bayesian clustering approach (STRUCTURE) proposed by Pritchard et al. (2000). For this analysis, we carefully excluded loci that were assumed to be null alleles and could therefore distort the results. Following this procedure, 37 loci were available for this analysis. We performed 10 independent runs (K=1–10) with 2 × 106 Markov chain Monte Carlo (MCMC) repetitions and a burn-in period of 50 000 iterations under an admixture ancestral model and a model in which allele frequencies were correlated. We calculated the posterior probability for each K value using the estimated log-likelihood of K to choose the optimal K.
We assessed the pattern of isolation by distance by comparing genetic distance against geographic distance between pairs of populations. The geographic distance between populations was considered to be the shortest distance between populations on the map. The significance of the association between the two types of distance was determined using the Mantel test (Mantel, 1967) using a permutation procedure with 10 000 re-samplings.
Relationship between FST values and heterozygosity
We also compared the distribution of the GST values over all loci to their expected values under the neutral assumption to detect nonneutral loci. In this analysis, we chose to use only codominant loci that showed Hardy–Weinberg equilibrium because we used loci that allowed us to estimate exact He values. Beaumont and Nichols (1996) showed that the distribution of FST as a function of heterozygosity in an island model is quite robust with respect to variations in conditions such as the population structure, demographic structure and mutation level. Therefore, we applied this method to identify markers deviating from the null hypothesis of neutral evolution. All GST values were transformed into FST values using the Cockerham and Weir (1987) transformation, FST=nGST/[GST+(n−1)], where n is the number of populations, and FST values were plotted as a function of the expected heterozygosity. This analysis was done in a two-step procedure (Beaumont and Nichols, 1996; Scotti-Saintagne et al., 2004), in which the first envelope for neutral expectation was estimated using the overall mean value of FST. Markers with FST values outside the 95% envelope were removed and a new analysis was done on the basis of the mean value of FST. Markers with FST values outside the 95% envelope after the second analysis were considered to be outliers. These calculations were done using Fdist2 software (Beaumont and Nichols, 1996).
Results
Characterization of CAPS markers
We were able to assign putative functions to 35/51 CAPS markers by BLAST search (Table 2). Two-thirds of our CAPS markers were probably based on low copy number genes, but 19 of them were highly redundant in the C. japonica database and the P. taeda, A. thaliana, and O. sativa databases (Table 2). Of the 51 loci, 10 showed dominant inheritance patterns (Table 2).
Genetic diversity and differentiation
The mean proportion of polymorphic loci (Pl) was 0.860, but the proportions for the two southern-most populations, Kobayashi and Yakushima, were lower than 0.800 (Table 1). The number of alleles per locus (Na) depended on the number of individuals investigated, but the latter did not vary greatly among populations (18–24 individuals), and we found few rare alleles in the CAPS analysis. Only three populations showed Na values lower than 1.90: the northernmost population at Iwaki, and the two southern-most populations at Kobayashi and Yakushima. These three populations were the only ones for which allelic richness (Rs) estimates were lower than 1.860. The average expected heterozygosity (He) of all populations was greater than 0.300, except for the two northern-most populations (Iwaki and Yaita), the Aokigahara population in north-central Japan, and the two southern-most populations (Kobayashi and Yakushima). The observed heterozygosity (Ho) did not completely follow the trend for He. The overall FIS values for the 51 loci were negative for each population, but not significantly different from those expected under Hardy–Weinberg equilibrium, except for those of the Chichibu, Tanakamiyama and Shiragayama populations. Thus, almost all of the populations had a slight excess of heterozygotic individuals. The number of loci with significant departures from Hardy–Weinberg equilibrium ranged from four in the Yaita population to 11 in the Miyamura, Tanakamiyama and Yakushima populations, and the pattern of deviation varied depending on the locus. All parameters of genetic diversity in the peripheral populations such as the Iwaki and Yaita populations in the north and the Kobayashi and Yakushima populations in the south tended to have significantly lower genetic diversity (Wilcoxon signed-ranks test after Bonferroni correction; P<0.05 and P<0.01 for He and Rs, respectively).
The allele frequencies at 10 loci in the populations showed a gradual cline with distance from the marginal population (CC0822, CC0860, CC1415, CC1463, CC1606, CC2478, CC2921, CC3162, CC3872 and CD0776), whereas at another seven loci (CC0530, CC0550, CC0951, CC1371, CC2750, CC2860 and CC3455) only some of the more remote populations at both the northern and southern ends of Japan had higher or lower frequencies than those of the more central populations.
Genetic structure and linkage disequilibrium
The overall genetic differentiation among populations at the 51 loci was low (GST=0.039), but the GST values varied among loci, ranging from 0.129 at locus CC2750 to 0.000 at loci CC0731, CC1172, CC2435 and CC2831 (Table 3). We also conducted AMOVA to determine the variation within and among populations and to test the significance of the among-population variation. The variation among populations was 0.0343 and was highly significant (P<0.001); it was also very similar to the overall GST value. The hierarchical AMOVA analysis revealed that the degrees of differentiation (FST) among groups and among populations within a group were 0.0396 and 0.0231, respectively.
Populations were generally clustered within the same district or group of districts, and thus showed a geographic trend in the neighbor-joining phylogenetic tree (Figure 2); the three main groupings included population in the Kanto and South Tohoku districts; populations in the Chugoku, Shikoku and Kyushu districts; and populations in the Chubu and Kinki districts. The positions of two populations (Tanakamiyama in Kinki district and Hikosan in Kyushu district) were not consistent with their geographical locations. Populations located within the main area of the species' natural distribution, such as those in Chubu district, were very closely related and have not diverged greatly from each other, but more remote populations such as those at Iwaki and Yaita in the north and Yakushima, Kobayashi and Tsutsuga in the south showed considerably more genetic divergence. The bootstrap value for each branch was not high, but some branches for more remote populations (Iwaki and Yaita; Yakushima, Kobayashi and Tsutsuga) and the highest-elevation population (Hijiri) were higher (Figure 2).

Phylogenetic tree for the 25 natural C. obtusa populations created using the neighbor-joining method. This method used a genetic distance matrix based on Nei's genetic distance (Nei, 1978); the scale bar at the bottom of the figure represents Nei's genetic distance. Values above the horizontal lines in the tree represent bootstrap probabilities. The right side of the diagram indicates the district names for each group of populations.
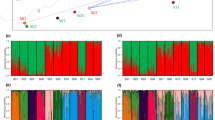
Bayesian clustering analysis of the 37 loci demonstrated that only the model with K=3 explained the data sufficiently well (this simulation had the highest log-likelihood value). This result suggests that the most probable number of populations was three, based on our data for 481 individuals from 25 populations. We genetically defined three groups by means of STRUCTURE analysis. The frequency of group 1 individuals (medium-gray in Figure 3) was high in populations within the Chubu district, which represents the main area of natural forest that currently exists. The frequency of group 3 (dark-gray in Figure 3) was relatively low in populations 1–13, increased from populations 14–22, and was especially frequent in the Shikoku and Chugoku districts. The frequency of group 2 (light-gray in Figure 3) was high in northern populations and declined as the prevalence of group 3 increased from populations 14–22.

Genetic relationships among the 25 populations surveyed in this study, including 481 individuals estimated from 37 CAPS loci using STRUCTURE (Pritchard et al., 2000).
We also investigated the association between the pairwise FST values and the geographical distances between populations, which represents the magnitude of isolation by distance (Figure 4). The association was highly significant (r=0.669, Mantel test, P<0.001); thus, isolation by distance was clearly a factor in the genetic structure of this species.

The relationship between geographical distance and the overall pairwise FST for 51 CAPS loci. The significance of this relationship, which represents the magnitude of the isolation by distance, was tested using the Mantel test (r=0.669, P<0.001).
Three populations (Tanakamiyama, Shiragayama and Yakushima) had high numbers of combinations of loci with significantly higher linkage disequilibrium values (P<0.05), 366, 342 and 422, respectively (Figure 5). The lowest value (124) was for the Yamazaki population, and the mean value was 235.

Linkage disequilibrium within each population. The values were calculated using the method of Weir (1990). The black and gray bars represent significance at P<0.01 and <0.05, respectively.
Relationship between FST values and heterozygosity
We used the overall mean FST value (0.048802) over 37 loci to construct the expected distribution of FST in an infinite-allele model (Figure 6). Seven loci were identified as outliers: CC0272, CC1172, CC1319, CC1625, CC2478, CC3872 and CC2750. CC2750 had the highest FST value; but the other six loci had very low FST values.

Distribution of FST values as a function of the within-population heterozygosity (HS) using 37 CAPS loci in C. obtusa. The envelope of values corresponds to neutral expectations (with FST=0.04) in the infinite-allele model constructed according to the method of Beaumont and Nichols (1996).
Discussion
Cleaved amplified polymorphic sequence markers for population study
We considered 21 CAPS loci to represent single loci that originated from single or low copy number genes because they showed only a single hit in the C. japonica database and no or only a few hits in the other databases (except CC1371 and CC2860). The nonredundant cDNA databases for C. japonica and C. obtusa do not seem to be saturated yet because of the limited number of clones they contain (∼15 000 and 5000 ESTs, respectively; Ujino-Ihara et al., 2005), but those of A. thaliana and O. sativa probably cover most of the genes in these species. The redundancy trend was similar in both the saturated and non-saturated databases. However, the CAPS patterns of highly redundant loci are simple and easily converted into genotypes. Inheritance modes of these CAPS markers have been confirmed by segregation analysis of artificially crossed populations (Matsumoto and Tsumura, 2004; Matsumoto et al., unpublished data). The average GST may have been underestimated when we included these loci with a dominant inheritance mode, but excluding these loci had little effect on calculated population genetic parameters, and the resulting genetic structure, of this species.
Genetic diversity between populations
According to our data, 10 of the 51 loci are likely to exhibit a genetic cline related to the geographic location of each population, a result that partly agrees with those of previous studies (Shiraishi et al., 1987; Uchida et al., 1997). Only the more peripheral populations at the southern and northern ends of Japan had significantly higher or lower frequencies, at seven loci, than those of the more central populations. These results probably reflect the expansion history of C. obtusa populations and migration between the central and more remote populations during the most recent postglacial period.
The more remote northern and southern populations of C. obtusa showed significantly lower genetic diversity. Therefore, we estimated the degree of genetic differentiation among three groups of populations: the remote northern populations (Iwaki and Yaita), the central populations, and the remote southern populations (Kobayashi and Yakushima). Our hierarchical AMOVA analysis revealed that the degree of differentiation (FST) among groups was 0.0396, very similar to that found in a previous study (GST=0.045; Uchida et al., 1997) and to the values reported for other conifers with similar ecological characteristics (Hamrick et al., 1992). Tajima (1990) showed that there should theoretically be a correlation between migration and DNA polymorphism in a local population; the expected amount of DNA polymorphism in a population with a lower migration rate is smaller than that of a population with a higher migration rate, suggesting that remote populations tend to have lower levels of DNA polymorphism than central populations when the migration rate to remote populations is lower than that to the central populations. The migration rate for the remote populations in our study is likely to be lower than that of the central populations because of the high GST values in remote populations, which consequently show a lower level of DNA polymorphism. Thus, our results confirm those of the theoretical study by Tajima (1990). Both the theoretical considerations and empirical data also suggest that the expansion of C. obtusa into more remote areas may not have been smooth, probably because at the southern end of the range there were many competitors during regeneration, such as non-deciduous trees (Takahara, 1998), while at the northern end the populations faced selective pressures exerted by the relatively severe winter climate.
Genetic structure of natural populations of C. obtusa
The genetic diversity of this species was slightly higher in populations in western Japan (with the exception of the Kyushu populations), which may have been a refugium during the last glacial period, than in populations in other areas.
The phylogenetic tree we constructed for C. obtusa was mostly consistent with the geographical location of each population (Figure 2), except for the Hikosan population, which clustered with the Tanakamiyama population and occupies a small area at the border of a large plantation of the same species. The allele frequencies of all loci in the Hikosan population were similar to those of the Tanakamisan population, and the results of our STRUCTURE analysis also mostly agree with this finding. Other studies (Uchida et al., 1997) have found the genetic differentiation between artificial populations to be very low compared with that between natural populations, and the artificial populations to be genetically closest to the natural populations in central Japan from the Kiso area to the Kii Peninsula. We carefully collected our study materials, but it is possible that some individuals were collected from artificial forests because the genetic diversity of the Hikosan population was also relatively high according to our results.
Our Bayesian analysis of the genetic structure defined three groups. However, the refugia of this species during the last glacial period remain unknown because of a lack of fossil pollen data, as described above. This species has probably undergone repeated distribution shrinkage and expansion as a result of climatic fluctuations since it first became established in Japan. Therefore, the origin of these three groups is not yet clear, but the findings suggest that at least three groups established and were subject to repeated episodes of genetic drift during these changes in distribution. The three groups may have migrated and exchanged genes between populations, leading to the current genetic structure of this species. Group 2 (light-gray in Figure 3) may have originated in the western populations, which are probably refugia for this species. In the near future, we may be able to confirm the genetic results if methods for analyzing fossil pollen become sufficiently precise to distinguish between pollen of the Cupressaceae, Taxaceae and Cephalotaxaceae.
The Tanakamiyama, Shiragayama and Yakushima populations had high numbers of combinations of loci with significant linkage disequilibrium values, which can be explained by recent genetic drift and subdivision of the populations. All populations of C. obtusa have experienced selective logging during the past. The Tanakamiyama population sustained particularly severe logging because it is close to historically large cities, such as Kyoto and Nara, and its timber was used heavily for construction in them. According to archeological data, the timber of this species accounted for about 60% of the total number of pillars used in the construction of castles and other large buildings in these cities (Shimachi and Ito, 1988). The Chamaecyparis forest of the Yakushima population has a very low density compared with the other forests, and this island is a World Heritage site that also contains Cryptomeria forest (Tsumura and Ohba, 1993). Since C. obtusa is rarely found in the Cryptomeria forests, we had to explore a large area to sample this population; consequently, we may have collected materials that resulted from nonrandom mating because of the very low density of C. obtusa. Human activities such as selective logging, combined with the low density of individuals in the population, may have been responsible for its significantly higher numbers of combinations of loci with significant linkage disequilibrium. These numbers may also have been influenced by aspects of the population's history, such as its date of establishment and the number of generations since the founder population (Hartl, 1980). This species also survived at least one glacial period, including the most recent glacial period from 20 000 to 15 000 years ago. During that ice age, all extant plant species in Japan had refugia in relatively warm areas such as the southwestern part of Japan, and then expanded their distribution northward during the postglacial period (Tsukada, 1983, 1988). The recently founded populations may still be too young to have attained linkage equilibrium because the generation time of this species is relatively long (several decades, on average). The total number of loci that we investigated may also have affected the linkage disequilibrium values because we used 51 loci in our study, and the basic chromosome number is 11 in this species; thus, some loci might be closely linked on the same chromosome. We are currently investigating the linkage among these loci using a mapping population. Eighteen of our 51 loci have been mapped in eight linkage groups, to date and some of them appear to be closely linked (Matsumoto et al., unpublished data).
The average FIS value indicates that the populations as a whole were not distorted from Hardy–Weinberg equilibrium, even though some loci were significantly distorted within populations. Three populations (Miyamura, Tanakamiyama and Yakushima) had the highest number of significant loci and the FIS values of most loci were negative in these populations. However, the Tanakamiyama population had positive values for six of these loci. Overexploitation of the natural forest followed by regeneration from a diminished population may be one reason for this finding.
Isolation by distance was demonstrated in the C. obtusa populations, especially for geographically close populations such as populations 7–13 in the Kiso area, in which this species has been intensively used to produce wood; these closely grouped populations appear to have diverged relatively little compared with more remote regions. These findings suggest that natural populations of C. obtusa still retain genetic structure in Japan, at least at the district level, and this structure can be seen in the phylogenetic tree that we created (Figure 2).
Outlier loci
We detected seven outlier loci, six of which had FST values lower than the 95% confidence interval. The FST values for these loci were fairly low (<0.01). Balancing selection (Strobeck, 1983; Hudson and Kaplan, 1988) may be responsible for these outlier loci if they confer advantages that are positively selected for in certain populations. In such cases, the some degree of polymorphism of the gene involved should be maintained in stable equilibrium, and the FST value would also be low. The copy numbers at three of these loci (CC0272, CC1172 and CC1625) was considered to be high (Table 2), suggesting that these markers are derived from multiple-copy genes. However, since there was either a low copy number or a single copy for three other loci (CC1319, CC2478 and CC3872), the outlier status of the genes may not be correlated with their copy numbers. The results of the homology search indicate that most of the corresponding genes may have ‘housekeeping’ functions, and their FST values may be low because of the importance of these functions. However, we were unable to determine the exact function of these genes in C. obtusa because the analysis was based on BLASTX search results. Thus, we must further investigate their functions. In contrast, CC2750 had the highest FST values, higher than the 95% confidence interval (Figure 4). The putative function of this gene is encoding a potassium channel tetramerization domain-containing protein, which functions as a voltage-gated potassium channel that senses changes in membrane potential and opens potassium-selective transmembrane pores, thereby regulating the electrical properties of cells (Bixby et al., 1999). However, there is no direct evidence regarding the role(s) of the gene in this species as yet. The four southernmost populations, except Hikosan, have unique allele frequencies of this gene, which is a candidate nonneutral gene and may play a specific role in local adaptation or differentiation.
The test to detect outlier loci for selection is unable to distinguish false positives (type I errors). Thus, we conducted the test with a 95% P level threshold, at which the expected number of loci for false positives is 2.55 (51 × 0.05), but we found only one outlier locus, suggesting that its candidate status must be treated with considerable caution (Vasemägi et al. 2005).
To obtain clear evidence for the adaptive value of this gene, we must analyze nucleotide variations in the gene between populations found in different environments.
Implications of the results for genetic resources of C. obtusa
We found that genetic differentiation within districts was relatively small, but the differentiation between districts was relatively large. In particular, more remote populations at both ends of the natural distribution (Iwaki and Yaita in the north and Yakushima and Kobayashi in the south) appear to have diverged, despite their relatively low genetic diversity. Genetic differentiation was much greater in western populations than in northern Japan, and the differentiation between populations in central Japan was very small. The small populations in western Japan and in Kanto district are more important, from a conservation perspective, than the populations in central Japan, owing to their genetic divergence, relatively small sizes and restricted areas. The largest trees in the natural forest have been used for construction of temples, shrines and modern houses. The current system of natural regeneration is designed to promote sustainable use of the forest (Yamamoto, 1998), but plantations of C. obtusa have increased and now account for 25% of the total area of artificial forest (10 million ha) in Japan. This means that pollen from the artificial forests will increasingly enter the natural forests and consequently affect the seedlings produced by natural regeneration. These seedlings will be contaminated because the supplied seedlings are from seed orchards (Moriguchi et al., 2005), which have been constructed from selected clones that originated mainly in central Japan. Thus, gene flow from the artificial forests will greatly modify the genetic structure of the natural populations, even when the clones have been selected from the same region as these natural forests; at a minimum, the genetic diversity will be decreased because the limited number of clones in the seed orchards represents a strong bottleneck for future generations. Fortunately, forest trees have long lives and can maintain their populations for several 100 years, even if the newly produced seedling populations cause gene contamination. Therefore, an ex situ conservation program is currently the best way to conserve the genetic diversity and structure of natural populations of C. obtusa. These conserved materials can be used as future sources of breeding materials and for the discovery of single nucleotide polymorphisms for important traits, because the species currently retains a clear genetic structure.
References
Acheré V, Favre JM, Besnard G, Jeandroz S (2005). Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol Ecol 14: 3191–3201.
Beaumont MA, Nichols RA (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond Ser B 263: 1619–1626.
Bixby KA, Nanao MH, Shen NV, Kreusch A, Bellamy H, Pfaffinger PJ et al. (1999). Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat Struct Mol Biol 6: 38–43.
Cockerham CC, Weir BS (1987). Correlations, descent measures: drift with migration and mutation. Proc Natl Acad Sci USA 84: 8512–8514.
Comps B, Gömöry D, Letouzey J, Thiebaut B, Petit RJ (2001). Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157: 389–397.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L. Skeels)] endemic to Morocco. Theor Appl Genet 92: 832–839.
Excoffier LP, Smouse E, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Felsenstein J (1995). PHYLIP (phylogeny inference package). Ver. 3.57c. Department of Genetics, University of Washington: Seattle.
Goudet J (2000). FSTAT: a program to estimate and test gene diversities and fixation indices. Ver. 2.9.1. Available viahttp://www2.unil.ch/popgen/softwares/fstat.htm.
Hamrick JL (2004). Response of forest trees to global environmental changes. For Ecol Manag 197: 323–335.
Hamrick JL, Godt MJW, Sherman-Broylers SL (1992). Factors influencing levels of genetic diversity in woody plant species. New Forests 6: 95–124.
Hartl DL (1980). Principles of Population Genetics. Sinauer Associates: Sunderland, MA.
Hayashi Y (1960). Taxonomical and Phytogeographical Study of Japanese Conifers. Norin-Shuppan: Tokyo (in Japanese).
Hudson RR, Kaplan NL (1988). The coalescent process in models with selection and recombination. Genetics 120: 831–840.
Lagercrantz U, Ryman N (1990). Genetic structure of Norway spruce (Picea abies): concordance of morphological and allozymic variation. Evolution 44: 38–53.
Lewis PO, Zaykin D (2002). GDA. Available viahttp://hydrodictyon.eeb.uconn.edu/people/plewis/software.php.
Mantel N (1967). The detection of disease clustering and a generalized regression approach. Can Res 27: 209–220.
Matsumoto A, Tsumura Y (2004). Evaluation of cleaved amplified polymorphic sequences markers for Chamaecyparis obtusa based on expressed sequence tag information from Cryptomeria japonica. Theor Appl Genet 110: 81–90.
Moriguchi Y, Tani N, Itoo S, Kanehira F, Tanaka K, Yomogida H et al. (2005). Gene flow and mating system in five Cryptomeria japonica D. Don seed orchards as revealed by analysis of microsatellite markers. Tree Genet Genome 1: 174–183.
Nei M (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70: 3321–3323.
Nei M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Nei M, Chesser RK (1983). Estimation of fixation indices and gene diversities. Ann Hum Genet 47: 253–259.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Saitou N, Nei M (1987). A neighbor-joining method: a new method for constructing phylogenetic trees. Mol Biol Evol 44: 406–425.
Schneider SJ, Kueffer M, Roessli D, Excoffier L (1997). Arlequin, Version 2.0: Software for Population Genetic Data Analysis. Genetic and Biometry Laboratory, University of Geneva: Switzerland. Available via http://lgb.unige.ch/arlequin/.
Scotti-Saintagne C, Mariette S, Porth I, Goicoechea PG, Barreneche T, Bodénès C et al. (2004). Genome scanning for interspecific differentiation between two closely related oak species [Quercus robur L. & Q. petraea (Matt.) Liebl.]. Genetics 168: 1615–1626.
Shimachi K, Ito T (1988). Comprehensive List of Unearthed Wood Products of Ruins in Japan. Yuzankaku-Shuppan: Tokyo (in Japanese).
Shimizu KK, Purugganan MD (2005). Evolutionary and ecological genomics of Arabidopsis. Plant Physiol 138: 578–584.
Shiraishi S, Kaminaka H, Ohyama N (1987). Genetic variation and differentiation recognized at two loci in hinoki (Chamaecyparis obtusa). J Jpn For Soc 74: 44–48.
Strobeck C (1983). Expected linkage disequilibrium for a neutral locus linked to a chromosomal arrangement. Genetics 103: 545–555.
Suzuki M (2002). Japanese and Wood Culture. Yasaka-Shobou: Tokyo (in Japanese).
Tajima F (1990). Relationship between migration and DNA polymorphism in a local population. Genetics 126: 231–234.
Takahara H (1998). Distribution history of Cryptomeria forest. In: Yasuda Y, Miyoushi N (eds). Vegetation History of the Japanese Archipelago. Asakura-Shoten: Tokyo (in Japanese). pp 207–223.
Tani N, Tomaru N, Araki M, Ohba K (1996). Genetic diversity and differentiation in populations of Japanese stone pine (Pinus pumila) in Japan. Can J For Res 26: 1454–1462.
Tomaru N, Mitsutsuji T, Takahashi M, Tsumura Y, Uchida K, Ohba K (1997). Genetic diversity in Fagus crenata (Japanese beech): influence of the distributional shift during the late-Quaternary. Heredity 78: 241–251.
Tsukada M (1983). Vegetation and climate during the last glacial maximum in Japan. Quaternary Res 19: 212–235.
Tsukada M (1988) Japan Handbook of vegetation science. In: Huntley B, Webb III T (eds). Vegetation History, Vol. 7. Kluwer: The Netherlands. pp 458–518.
Tsumura Y (2006). The phylogeographic structure of Japanese coniferous species as revealed by genetic markers. Taxon 55: 53–66.
Tsumura Y, Ohba K (1993). Genetic structure of geographical marginal populations of Cryptomeria japonica. Can J For Res 23: 859–863.
Tsumura Y, Suyama Y (1998). Differentiation of mitochondrial DNA polymorphisms in populations of five Japanese Abies. Evolution 52: 1031–1042.
Tsumura Y, Yoshimura K, Tomaru N, Ohba K (1995). Molecular phylogeny of conifers using RFLP analysis of PCR-amplified specific chloroplast genes. Theor Appl Genet 91: 1222–1236.
Uchida K, Tomaru N, Tomaru C, Yamamoto C, Ohba K (1997). Allozyme variation in natural populations of hinoki, Chamaecyparis obtusa (Sieb. et Zucc.) Endl. and its comparison with the plus-trees selected from artificial stands. Breed Sci 47: 7–14.
Ujino-Ihara T, Kanamori H, Yamane H, Taguchi Y, Namiki N, Mukai Y et al. (2005). Comparative analysis of expressed sequence tags of conifers and angiosperms reveals sequences specifically conserved in conifers. Plant Mol Biol 59: 895–907.
Vasemägi A, Nilsson J, Primmer CR (2005). Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in atlantic salmon (Salmo salar L.). Mol Biol Evol 22: 1067–1076.
Weir BS (1979). Inferences about linkage disequilibrium. Biometrics 25: 235–254.
Weir BS (1990). Genetic Data Analysis. Sinauer Associates: Sunderland, MA.
Wright S (1922). Coefficients of inbreeding and relationship. Am Natur 56: 330–338.
Yamamoto S (1998). Regeneration ecology of Chamaecyparis obtusa and C. pisifera (hinoki and sawara cypress), Japan. In: Laderman AD (ed). Coastally Restricted Forests. Oxford University Press: Oxford, UK. pp 101–110.
Acknowledgements
We express our sincere thanks to H Tachida for insightful comments and discussion concerning earlier versions of this manuscript and H Takahara for helpful comments regarding the fossil pollen data. We also thank the staff at each of the following: Chichibu University Forest (University of Tokyo), Ashiu University Forest (Kyoto University), Wakayama University Forest (Hokkaido University), Sumitomo Forestry Co Ltd, Tokai-Pulp and Paper Co Ltd, Senshusha Co Ltd, Tsutsuga Village Office, and Regional Forest Offices for allowing us to collect samples, and M Koshiba and Y Taguchi for their excellent technical assistance. This study was partly supported by the Japan Society for the Promotion of Science, Grant-in-Aid for Scientific Research (B), No. 16380112.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsumura, Y., Matsumoto, A., Tani, N. et al. Genetic diversity and the genetic structure of natural populations of Chamaecyparis obtusa: implications for management and conservation. Heredity 99, 161–172 (2007). https://doi.org/10.1038/sj.hdy.6800978
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6800978
Keywords
This article is cited by
-
The origin and genetic variability of vegetatively propagated clones identified from old planted trees and plantations of Thujopsis dolabrata var. hondae in Ishikawa Prefecture, Japan
Tree Genetics & Genomes (2019)
-
Detecting east–west genetic differentiation in Castanopsis (Fagaceae) on the main islands of Japan and north–south on the Ryukyu Islands, based on chloroplast haplotypes
Plant Systematics and Evolution (2016)
-
Twenty-three years of stand dynamics in an old-growthChamaecyparisforest in central Japan
Journal of Forest Research (2014)
-
Phylogeographic patterns of highland and lowland plant species in Japan
Alpine Botany (2011)
-
Genetic diversity and structure of natural fragmented Chamaecyparis obtusa populations as revealed by microsatellite markers
Journal of Plant Research (2010)
