
Abstract
This study used allozyme and mitochondrial DNA variation to examine genetic structure in the Oxleyan Pygmy Perch Nannoperca oxleyana. This small-bodied freshwater fish has a very restricted distribution occurring only in some small coastal streams in south-east Queensland and northern New South Wales. It was expected that subpopulations may contain little genetic variation and be highly differentiated from one another. The results, based on allozyme and mitochondrial DNA control region variation were in agreement with these expectations. Allozyme variation was very low overall, with only one locus showing variation at most sites. The high differentiation was because a different locus tended to be polymorphic at each site. Mitochondrial variation within sites was also low, but some sites had unique haplotypes. The patterns of similarity among mitochondrial DNA haplotypes were not as expected from geographical proximity alone. In particular, although some northern sites had unique haplotypes, four sites spread along 200 km of coastline were remarkably similar, sharing the same common haplotype at similar frequencies. We suggest that these four streams may have had a confluence relatively recently, possibly when sea levels were lower, 8000–10 000 BP.
Similar content being viewed by others

Introduction
The role that current and historical patterns of dispersal have in population genetic structure of natural populations has been assessed for a range of terrestrial and aquatic species (for example, Reeb & Avise, 1990; Templeton et al., 1995). In general, species that have efficient mechanisms for dispersal show limited genetic structuring of their subpopulations, whereas in those species where dispersal is restricted substantial differentiation among subpopulations may occur (Slatkin, 1981). In a review of published studies on marine, anadromous and freshwater fishes, Ward et al. (1994) concluded that generally marine fish species exhibit the lowest levels of genetic variation among subpopulations, but the highest levels within subpopulations. In contrast, freshwater species tend to exhibit high levels of structure among subpopulations and low levels of variation within subpopulations. These differences are thought to be largely the result of the differing potential for dispersal among subpopulations of marine and freshwater species. Many freshwater species are restricted to their particular drainage, and dispersal among drainages is likely to be limited by the marine sections separating them. Because the subpopulations are relatively isolated from one another, they will tend to diverge as a result of genetic drift and/or selection. The rate of divergence will be affected by the effective population size and the strength of any selection pressures. Small populations under intense selection would be expected to diverge more quickly than large populations under minimal selection. Thus for those freshwater species that are rare or threatened, that live in specific habitats and usually in small populations, low levels of genetic variation may be expected within populations, whereas substantial differentiation between populations from different drainages may be expected.
The Oxleyan Pygmy Perch Nannoperca oxleyana (family Nannopercidae) inhabits small coastal streams on the mainland and islands of south-eastern Queensland and a few localities in northern New South Wales, Australia (Merrick & Schmida, 1984). The Action Plan for Australian Freshwater Fishes (Wager & Jackson, 1993) classified the Pygmy Perch as ‘vulnerable’ because of its restricted geographical distribution, its absolute dependence on dystrophic waterways of low pH associated with heath vegetation, and low population sizes in many localities. Although N. oxleyana is not presently ‘endangered’, most of its known populations are threatened to some degree by habitat fragmentation, water pollution, collecting for aquarium purposes or by the exotic mosquitofish Gambusia holbrooki (Arthington & Marshall, 1996; Kuiter et al., 1996). In developing a recovery plan for the Pygmy Perch (Arthington, 1996), it was considered prudent to determine the genetic structure of the remaining populations and to use this information to maximize the amount of genetic diversity to be maintained through conservation measures.
Many previous studies have used allozyme analysis to assess the levels and patterns of variation among subpopulations of different species (e.g. fish, Meffe & Vrijenhoek, 1988; Crustacea, Hughes et al., 1995; insects, Schmidt et al., 1995). This technique allows inferences to be drawn about the level and pattern of dispersal among subpopulations and their degree of isolation. More recently, variation in mitochondrial DNA has also been used for examining population structure. Mitochondrial DNA is generally maternally inherited in vertebrates and is haploid (Avise, 1991). Thus mitochondrial genes have effective population sizes roughly one quarter those of nuclear genes (Birky et al., 1989). This means that mtDNA is affected more by genetic drift than is nuclear DNA, resulting in a more rapid loss of diversity within small isolated subpopulations and a more rapid rate of differentiation among subpopulations. As a result, the analysis of mtDNA has the potential to be more sensitive than nuclear DNA markers for detecting limited dispersal among subpopulations, although this is not always the case (e.g. Ward & Grewe, 1995 and references therein).
The primary aim of this study was to use variation in both nuclear genes (allozymes) and mtDNA to assess the amount of genetic variation within and among populations of the Pygmy Perch. Given its limited distribution and relatively low densities, we expected low levels of variation within subpopulations from particular drainages and high levels of differentiation among drainages. The second aim was to define the implications of the results for the long-term conservation of N. oxleyana.
Materials and methods
Sample collection
Fish were collected from nine sites in south-eastern Queensland, which covered almost all the species’ range. We were unable to obtain samples from northern New South Wales as the species is very uncommon there. There were three sites on Moreton Island, one on Fraser Island and five on the adjacent mainland. Precise localities are shown in Fig. 1. The Noosa River sample included fish collected from six different sections of the river. The other samples were all collected from small streams. Seine nets were run within 3 m of stream bank and around macrophyte beds. Where possible 30 fish were sampled from each site. This usually entailed sampling along a substantial stretch of stream, up to 100 m, because fish were not common in most streams. A sample size of 30 individuals provides a 95% chance of detecting at least one copy of each mtDNA haplotype that occurs at 10% or greater frequency in the population and a 95% chance of detecting alleles at allozyme loci that occur at 5% or greater frequency (Schwager et al., 1990). Fish were frozen in liquid nitrogen and transported back to the laboratory where they were stored at −75°C until needed for genetic analysis.

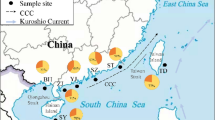
Map of south-east Queensland coast, showing sampling sites for Pygmy Perch, pie diagrams indicating mtDNA haplotype frequencies at each site and a network indicating relationships between haplotypes.
Allozyme analysis
A small sample of muscle tissue was removed from each fish and ground by hand in 0.2 mL of buffer (2.44 g Trizma base, 0.37 g EDTA free acid, 5.36 g NH4Cl, 19.8 g glucose, 20 mL 0.022 M NaNO3, in 1 L of H2O). Samples were centrifuged at 16 800 g for 20 min in a Sorvall RC5B refrigerated centrifuge operating at 5°C. Electrophoresis was performed on cellulose acetate plates (Titan III, Helena Laboratories). A total of 30 enzymes was screened, of which 23 gave clearly interpretable banding patterns. Only six loci were polymorphic. These were phosphoglucoisomerase (PGI, EC no. 5.3.1.9), phosphoglucomutase (PGM, EC no. 5.4.2.2), aspartate aminotransferase (AAT, EC no. 2.6.1.1), lactate dehydrogenase (LDH, EC no. 1.1.1.27), esterase (EST, EC no. 3.1.1.1) and alcohol dehydrogenase (ADH, EC no. 1.1.1.1). Unfortunately not all individuals could be scored for β-Est and Adh, so that sample sizes differ among loci for some sites (Appendix 1).
Analysis of mitochondrial DNA
Total DNA was extracted, by homogenizing ≈25 mg of white muscle tissue in 350 μL of extraction buffer (0.5 M Tris 8.0; 0.25 M NaCl, 0.025 M EDTA; 0.175 mM SDS) and incubated at 55°C overnight. Proteins and lipids were removed by sequential extractions with equal volumes of phenol–chloroform–isoamyl alcohol (25 : 24 : 1) and chloroform–isoamylalcohol (24 : 1). DNA was precipitated at 4°C in the presence of isopropanol (1 vol.). Pellets were washed in 1000 μL of ethanol (70%), dried and resuspended in 50 μL of TE buffer (10 mM Tris HCl, 1 mM EDTA, pH 7.5).
Two oligoprimers were used to amplify a 450-bp fragment of the mitochondrial control region. The heavy strand primer (MT16498H; 5′-CCT GAA GTA GGA ACC AGA TG-3′; (Meyer et al., 1990) is located in a conserved region within the control region. The light strand primer (L19; 5′-ACC ACT AGC ACC CAA AGC TA-3′; Bernatchez & Danzmann, 1993) is in the proline tRNA gene which flanks the 3′ end of the control region.
PCR reactions contained 30 nmoles each of dATP, dGTP, dCTP and dTTP (Promega), 1 unit of Taq DNA polymerase (Promega), 2.5 μmoles of MgCl2, 0.5 μmoles of each primer, 5 μL of 10× polymerase reaction buffer (Promega), 0.2 μg of template DNA, adjusted to final volume of 50 μL with ddH2O and overlayed with 30 μL of light mineral oil. Mixtures were then subjected to 35 cycles of: 5 min at 94°C (only on initial cycle), 30 s at 94°C, 30 s at 50°C, 1 min at 72°C, 5 min at 72°C (final cycle only). Samples were stored at 4°C.
Temperature gradient gel electrophoresis combined with heteroduplex analysis (TGGE-HA) was used to screen amplified fragments for variation. Prior to running the TGGE, samples were heteroduplexed with a reference conspecific. Samples contained 10 ng of reference conspecific and sample amplified DNA, 4 L. urea and 1× ME (20 mL. MOPS, 1 mL. EDTA) in a total volume of 5 μL. Each mixture was brought to 94°C for 5 min (for denaturation), then 50°C for 14 min (for heteroduplex formation) and allowed to equilibrate at room temperature for 10 min before being loaded on to the gel. The gel was a 5% denaturing polyacrylamide gel (8 L. urea, 2% glycerol, 30 : 0.5 acrylamide:bis in 1× ME buffer). Gels were run through a temperature gradient of 17–52°C at 300 V for 3.5 h.
Gels were silver stained as recommended by the manufacturers (QIAGEN). All samples were run with the same reference individual for heteroduplex analysis. This was then repeated with an individual of a different haplotype, to ensure that gels were being interpreted correctly. No discrepancies were found in assigning haplotypes.
Of the putative haplotypes identified by TGGE-HA, one individual of each haplotype from each site was sequenced. PCR product was cleaned using Quiaquick columns (Quiagen) following the manufacturers specifications. Sequencing reactions were carried out using dye terminator cycle sequencing reactions (Perkin Elmer) according to manufacturers instructions along with ≈30 ng of clean DNA template and 3.2 pmol of primer on an Applied Biosystems 373 automated sequencing machine. Both light and heavy strands were sequenced.
Statistical analysis
For the allozyme data, L. 1.7 (Swofford & Selander, 1989) was used to calculate allele frequencies, percentage polymorphism and average heterozygosity for each site, and to test for deviations from Hardy–Weinberg equilibrium. Wright’s F-statistics were used to assess population structure. Further analyses were carried out to examine variation within regions (i.e. among sites on Moreton Island and among mainland sites from Searys Creek South to Mellum Creek) and between regions. The regions were (a) Fraser Island (b) Coondoo Creek (c) Mainland south: Searys Creek, Noosa River, Marcus Creek and Mellum Creek, and (d) Moreton Island: Lake Jabiru, Spitfire Creek and Tempest Creek. Coondoo Creek was analysed as a separate region because it enters the sea via the Mary River, a long way further north than Searys Creek.
Haplotype diversity was calculated for each subpopulation (Nei, 1987). A minimum-spanning network of the evolutionary relationships among mtDNA haplotypes was constructed with the assistance of a program supplied by L. Excoffier. The groups of closely related haplotypes defined by the network determined our groupings for the subsequent population genetic analyses. The degree of population subdivision was estimated using a nested Analysis of Molecular Variance (L.) (Excoffier et al., 1992). Groups for the analysis were the same as for the allozyme analysis. This technique partitions variation according to sampling design and yields Φ-statistics which are analogues of Weir & Cockerham’s (1984) FST estimator, θ, when haplotype frequencies only are used in the analysis. L.s were performed both on haplotype frequencies alone and using the sequence divergence among haplotypes assuming a Tamura & Nei (1993) gamma model of evolution (gamma parameter=0.5) to correct for unequal nucleotide substitution among sites. Φ-statistics were tested for significance using 1000 random permutations of the original data (Schneider et al., 1997).
Results
Allozymes
FIS values were all nonsignificant, with the exception of Pgm-1 in the Noosa River (Table 1). This could have occurred because this sample was a combination of samples from separate sections of the river. The levels of variation within populations were very low (Table 2), with 1.1 alleles per locus, percentage polymorphism between 0.0 and 14.8% and an average heterozygosity of 0.016. In most populations, only a single locus was polymorphic, but different loci were polymorphic in different populations. This resulted in an FST value of 0.211, i.e. 21.1% of the total variation was among subpopulations. Lake Jabiru and Spitfire Creek were very similar, as may be expected because they are currently connected within a drainage system (Table 3). The variation among sites on Moreton Island was less than among sites on the mainland, but was still highly significant at three loci (Table 4, P < 0.001).
A total of seven mitochondrial DNA haplotypes was identified (Fig. 1). The variation within populations was again low. Eight of the nine populations were either totally monomorphic or were dominated by a single haplotype. Only one population, from Marcus Creek on the mainland, showed a second haplotype at a frequency of more than 0.25. The haplotype diversity within populations was also low, ranging from 0.00 to 0.47 (Table 5). Differentiation among populations was extremely high, with an FST of 0.87, i.e. 87% of the total variation was between subpopulations. The L. based on haplotype frequency indicated that the genetic variation was much greater among the broad geographical groupings than within them (Table 6). The result was very similar when sequence divergence was included. The three subpopulations from Moreton Island were very similar, all sharing the same haplotype at high frequencies (Fig. 1). The pairwise FST analysis indicated that they were not significantly different (Table 5, P > 0.05). The other populations that appeared very similar were those found in coastal streams from Searys Creek South to Mellum Creek. Again the same haplotype was most common in all these sites, although some pairs of sites did differ significantly in frequency of haplotypes (Fig. 1, Table 7). The samples from Coondoo Creek in the Mary River catchment and Coongul Creek on Fraser Island were different from each other and from all other samples, each being fixed for a unique haplotype.
Although the network illustrating relationships among the seven haplotypes is very shallow, with only four or five base pair substitutions separating the most divergent haplotypes, there is still a clear geographical pattern (Fig. 1). All three haplotypes on Moreton Island form a single clade. The haplotype that is common in coastal streams up to Searys Creek is central to the network and each of the other haplotypes found in these streams differ from it by only a single base pair substitution. The Fraser Island haplotype also differs from the common one by a single base pair and the Coondoo haplotype is one further base pair removed again.
Discussion
The results from the analysis of allozyme variation suggest very low levels of heterozygosity within subpopulations. These levels are low even for freshwater fish, which according to Ward et al. (1994) have average heterozygosity levels around 0.046. Of the 49 species reported in their summary, only three species had levels lower than reported here for N. oxleyana. Ward et al. (1994) report heterozygosity levels of 0.052 for anadromous fishes and 0.059 for fully marine species. Our result is not surprising given the degree of isolation of N. oxleyana populations, tending to accentuate the effects of genetic drift, whereas any loss of variation resulting from drift in more connected populations will tend to be counteracted by gene flow. Surprisingly the Tempest Creek population had the highest level of heterozygosity, even though it was one of the smallest streams sampled.
Similarly low levels of intrapopulation diversity were seen in the mtDNA. This is more difficult to compare with other published work, because results depend on the particular fragment of the mitochondrial genome that is being analysed. The control region is recorded as having the greatest amount of variation in most species (Avise, 1991) but even different parts of that region may vary in the amount of variation within populations (Faber & Stepien, 1997). Chenoweth & Hughes (1997) examined variation in the same section of the mitochondrial genome in the Australian bass (Macquaria novomaculeata), a catadromous species. Much higher levels of variation were found within populations (within population diversity values of 0.587). The same fragment has also been examined in the Barramundi (Lates calcarifer), a catadromous species thought to migrate at least between adjacent river systems. Extremely high levels of variation were found (Chenoweth et al., 1998). These differences are not unexpected as both the bass and the barramundi are likely to have much larger effective population sizes than the Pygmy Perch, which is restricted to particular freshwater habitats, occurs mainly in low densities and cannot disperse through salt water (Kuiter et al., 1996).
The levels of variation among populations of N. oxleyana are also quite high. Twenty-one percent of the allozyme variation and 87% of the control region variation was distributed among subpopulations. These results support the idea that dispersal among populations is extremely limited. The higher levels of differentiation for mtDNA were not unexpected, as mtDNA is expected to differentiate at a faster rate than nuclear DNA, because of its maternal inheritance and haploid nature and thus lower effective population size (Birky et al., 1989).
On the basis of the allozyme data alone, it could be suggested that the only two subpopulations likely to exchange individuals currently are Lake Jabiru and Spitfire Creek. This is not surprising, as these two sites are currently connected via a swampy drainage system. Significant differences in allele frequencies are seen between all other pairs of sites suggesting no or very limited dispersal of fish between them. This may seem an obvious result as the subpopulations came from different streams. However, during flood conditions it was considered possible that some areas of coastline would be fresh, thus allowing the possibility of dispersal between nearby streams.
The mtDNA results suggest a similar pattern, particularly with respect to Mary River and more southern coastal rivers, and the fixed differences between island and mainland sites indicate no recent dispersal of fish between them. The similarities among the four streams from Searys south to Mellum Creek could be interpreted either as the result of some current gene flow between these sites or the retention of variation arising from relatively recent connection of these rivers. The first interpretation seems unlikely, given the sensitivity of this species to brackish or saline water (Kuiter et al., 1996) and the fact that the allozyme data indicate limited gene flow. The other alternative, that these rivers have been connected relatively recently is entirely possible. Sea level changes over the late Pleistocene have been extensive (Chappell & Shackleton, 1986) and as recently as 8000 years ago, the coastline would have extended out past both Fraser and Moreton Island. Our data could indicate that at this time, these four streams had a confluence before reaching the sea. Although no published studies exist on this area, information from the Queensland Department of Minerals and Energy (A. Stevens, pers. comm.) indicates the possibility that the Brisbane River, which currently flows out through Moreton Bay, may have flowed a long way further north before entering the sea. All mainland streams south of Fraser Island may have flowed into the Brisbane River prior to entering the sea and thus formed part of the same drainage. This suggestion is purely speculative and, because of the high wave action on that particular stretch of coastline, even seismic profiling is unlikely to reveal the positions of past drainages (A. Stevens, pers. comm.).
That the three haplotypes on Moreton Island form part of the same clade suggests that at least the two rare ones have arisen since these sites became separated from the mainland streams. Although Moreton Island was attached to the mainland as recently as 8000 L., evidence from the mtDNA data suggests that the Moreton Island streams may not have connected with easterly flowing streams along the Sunshine Coast at that time, as indicated by the greater similarity of Moreton Island haplotypes to each other than to any mainland haplotypes.
Similarly, mitochondrial DNA data from Fraser Island and Coondoo Creek suggest that these populations have been separated from the other mainland populations for longer than the other mainland populations have been isolated from each other, because they have no haplotypes in common and appear to form a separate clade. This is not surprising as geomorphological evidence suggests that a ridge separates the Mary and southern drainages (Bridges et al., 1990). The larger differences between the two northern sites, compared to the four other mainland sites, also suggest that in recent times, Searys Creek, which flows into Tin Can Bay, has flowed out to the south, rather than north of Fraser Island, via the Mary River (Fig. 1).
The evolutionary sequence resulting in the present-day patterns can be inferred from the congruence of the network with geographical distribution. The most likely scenario is that the present haplotypes evolved from a common ancestor, probably the central haplotype, that was widely distributed. The base substitutions probably occurred once the populations had become isolated from one another, possibly with the Coondoo Creek haplotype evolving later from the Fraser Island one. The alternative, that all haplotypes were present at the time of isolation and different ones have become dominant (or fixed) since, is less likely because the geographical concordance with the network would not be expected.
Interestingly, a recent study of genetic variation in a freshwater shrimp, which inhabits the same coastal heath streams as the Pygmy Perch, has indicated very similar patterns (Woolschot, 1997). Allozyme variation within and between populations from mainland streams shows that subpopulations from the Noosa River right down to rivers opposite Moreton Island are very similar in allozyme frequencies, but highly significantly different from those flowing into Coondoo Creek in the Mary River drainage (FST values=0.65).
As the geomorphological evidence for river confluence is inconclusive, we plan to examine a number of other freshwater species from this region, that may be expected to have experienced the same historical events of isolation and connection. Similar patterns of genetic similarity would be expected, if the dispersal abilities and recent histories of the species coincide.
The implications of these results for the long-term conservation of N. oxleyana are significant. Most of the genetic diversity in the species is found between subpopulations from isolated localities rather than within subpopulations. Therefore, to maximize the preservation of genetic diversity as many subpopulations as possible should be maintained. In addition the large differences between subpopulations indicate that they have been isolated from one another for a considerable period of time. This should be taken into account if any restocking from other subpopulations or from stock bred in aquaria is to be considered. Finally, it is clear that the dispersal abilities of this species are very limited and that isolated subpopulations, once extinct are likely to remain so.
A number of fish and crustaceans have distributions and habitat requirements similar to N. oxleyana in south-east Queensland. If they cannot disperse among streams at flood time, as seems to be the case of N. oxleyana, it seems likely that the subpopulations in particular streams will be genetically differentiated to a similar extent. We are currently examining two additional heathland fish species, a threatened species and a common widespread species. Both are restricted to fresh water and occur in the same geographical area as N. oxleyana. This may shed light on whether the strong structure in N. oxleyana is largely a result of small population sizes or whether the limits to dispersal alone are more likely to have led to this pattern.
References
Arthington, A. H. (ed.) (1996). Recovery Plan for the Oxleyan Pygmy Perch Nannoperca oxleyana. Final Report to the Australian Conservation Agency, Canberra, CT.
Arthington, A. H. and Marshall, C. J. (1996). Threatened fishes of the world: Nannoperca oxleyana Whitley, 1940 (Nannopercidae). Environ Biol Fishes. 46: 150–150.
Avise, J. C. (1991). Ten unorthodox perspectives on evolution prompted by comparative population genetic findings on mitochondrial DNA. Ann Rev Genet, 25: 45–69.
Bernatchez, L. and Danzmann, R. G. (1993). Congruence in the control-region sequence and restriction-site variation in mitochondrial DNA of brook char (Salvelinus fontinalis Mitchell). Mol Biol Evol, 10: 1002–1014.
Birky, C. W., Fuerst, P. and Maruyama, T. (1989). Organelle gene diversity under migration, mutation, and drift: equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics. 21: 613–627.
Bridges, E. M., Ross, D. J. and Thompson, C. H. (1990). Soils of the Mary River Alluvia near Gympie, Queensland. CSIRO Division of Soils, Division Report No. 109.
Chappell, J. and Shackleton, N. G. (1986). Oxygen isotopes and sea level. Nature. 324: 137–140.
Chenoweth, S. and Hughes, J. M. (1997). Genetic population structure of the catadromous perciform Macquaria novomaculata (Percichthyidae). J Fish Biol, 50: 721–733.
Chenoweth, S., Hughes, J. M., Keenan, C. P. and Lavery, S. (1998). Concordance between dispersal and mitochondrial gene flow: isolation by distance in a tropical teleost, Lates calcarifer (Australian barramundi). Heredity. 80: 187–197.
Excoffier, L., Smouse, P. E. and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 131: 479–491.
Faber, J. E. and Stepien, C. A. (1997). The utility of mitochondrial DNA control region sequences for analysing phylogenetic relationships among populations, species and genera of the Percidae. In: Kocher, T D and Stepien, C A (eds). Molecular Systematics of Fishes, pp. 129–143. Academic Press, San Diego CA.
Hughes, J. M., Bunn, S. E., Kingston, M. and Hurwood, D. A. (1995). Genetic differentiation and dispersal among populations of Paratya australiensis (Atyidae) in rainforest streams in southeast Queensland, Australia. J N Am Benthol Soc, 14: 158–173.
Kuiter, R. H., Humphries, P. and Arthington, A. H. (1996). Pygmy Perches — Nannopercidae. In: McDowell, R. M. (ed.). Freshwater Fishes of South-Eastern Australia, pp. 168–175. Reed, Sydney.
Meffe, G. K. and Vrijenhoek, R. C. (1988). Conservation genetics in the management of desert fishes. Conserv Biol, 2: 157–169.
Merrick, J. R. and Schmida, G. E. (1984). Australian Freshwater Fishes: Biology and Management. Griffin Press, Adelaide.
Meyer, A., Kocher, T. D., Basasibawaki, P. and Wilson, A. C. (1990). Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNA sequences. Nature. 347: 550–553.
Nei, M. (1987). Molecular Evolutionary Genetics. Columbia University Press, New York.
Reeb, C. A. and Avise, J. C. (1990). A genetic discontinuity in a continuously distributed species: mitochondrial DNA in the American Oyster, Crassostrea virginica. Genetics. 124: 397–406.
Schmidt, S. K., Hughes, J. H. and Bunn, S. E. (1995). Gene flow among conspecific populations of Baetis sp. (Ephemeroptera): adult flight and larval drift. J N Am Benthol Soc, 14: 147–157.
Schneider, S., Kueffer, J., Roessli, D. and Excoffier, L. (1997). Arlequin: Exploratory molecular population genetics software, version 1. University of Geneva.
Schwager, S. J., Mutchler, M. A., Federer, W. T. and Scully, B. T. (1990). The Effect of Linkage on Sample Size Determination for Multiple Trait Selection. Tech. Report No. BU-1031-MA. Biometrics Unit, Cornell University, Ithaca, NY.
Slatkin, M. (1981). Estimating levels of gene flow in natural populations. Genetics. 95: 323–335.
Swofford, D. L. and Selander, R. B. (1989). BIOSYS-1. A computer program for the analysis of allelic variation in population genetics and biochemical systematics. Release 1.7. University of Illinois, Champaigne, IL.
Tamura, K. and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol, 10: 512–526.
Templeton, A. R., Routman, E. and Phillips, C. A. (1995). Separating population structure from population history: a cladistic analysis of the geographical distribution of mitochondrial DNA haplotypes in the Tiger Salamander, Ambystoma tigrinum. Genetics. 140: 767–782.
Wager, R. and Jackson, P. D. (1993). The Action Plan for Australian Freshwater Fishes Australian Nature Conservation Agency, Canberra.
Ward, R. and Grewe, P. M. (1995). Appraisal of molecular genetic techniques in fisheries. In: Carvalho, G. P. and Pitcher, J. J. (eds) Molecular Genetics in Fisheries Chapman & Hall, London.
Ward, R., Woodwark, M. and Skibinski, D. O. F. (1994). A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol, 44: 213–232.
Weir, B. S. and Cockerham, C. C. (1984). Estimating F -statistics for the analysis of population structure. Evolution. 38: 1358–1370.
Woolschot, L. (1997). Gene Flow and Dispersal Among Populations of the Freshwater Shrimp Caridina indistincta, in Lowland Coastal Streams of South-East Queensland. Honours Thesis, Griffith University.
Author information
Authors and Affiliations
Corresponding author
Appendix 1
Appendix 1
(See Table below)
Rights and permissions
About this article
Cite this article
Hughes, J., Ponniah, M., Hurwood, D. et al. Strong genetic structuring in a habitat specialist, the Oxleyan Pygmy Perch Nannoperca oxleyana. Heredity 83, 5–14 (1999). https://doi.org/10.1038/sj.hdy.6885390
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/sj.hdy.6885390
Keywords
This article is cited by
-
Characterizing the roles of changing population size and selection on the evolution of flux control in metabolic pathways
BMC Evolutionary Biology (2017)
-
Population genetics of a widely distributed small freshwater fish with varying conservation concerns: the southern purple-spotted gudgeon, Mogurnda adspersa
Conservation Genetics (2016)
-
Genetic diversity and population structure of the threatened freshwater catfish, Tandanus tandanus, in Victoria, Australia
Conservation Genetics (2015)
-
Urban development explains reduced genetic diversity in a narrow range endemic freshwater fish
Conservation Genetics (2015)
-
Evolution and maintenance of divergent lineages in an endangered freshwater fish, Macquaria australasica
Conservation Genetics (2010)
