
Abstract
In order to extend present knowledge of brown trout phylogeography in the Palaearctic, we analysed the complete mitochondrial D-loop sequence (1025–1027 bp) of all mitochondrial haplotypes of Salmo trutta found in the Iberian Peninsula and one North African haplotype. Sequence variation in the mitochondrial control region serves to identify four major haplotype groups within the Iberian Peninsula, i.e. Atlantic, Duero, Mediterranean and Andalusian. Including the Iberian haplotypes, the five main European groups previously established were increased to six: (i) an Atlantic group including two different clusters, South European and North Atlantic; (ii) a group representing an endemism restricted to the Duero basin in the Iberian Peninsula; (iii) an Adriatic–Andalusian group found in two vicariant areas including Adriatic–Ionian populations in the Mediterranean and the Andalusian basins of the southern Iberian Peninsula; (iv) a Mediterranean group with a distribution range that extends from the southwestern basins of the Iberian Peninsula to the Ionian basins of the Greek Peninsula; (v) a Danube group of wide distribution in the Black, Aral and Caspian basins; and (vi) a group comprising the S. t. marmoratus subspecies confined to the Adriatic Sea. The Iberian Peninsula appears to have acted as a physical boundary between haplotypes corresponding to Atlantic- and Mediterranean-draining rivers. Owing to its geographical position, this area has played a major role in present Palaearctic species distribution, as illustrated by its haplotype diversity.
Similar content being viewed by others

Introduction
Salmo trutta is one of the most important river fish species in Europe, in social, ecological and economic terms. It is distributed widely across Europe and is present in North Africa and western Asia, constituting a complex mosaic of phenetic variation (Behnke, 1972, 1986). The local genetic structure of brown trout has been investigated throughout Europe (Ferguson, 1989; Hamilton et al., 1989; García-Marín et al., 1999). Electrophoretic studies have defined ‘modern’ (North Atlantic) and ‘ancient’ (South Atlantic and Mediterranean) lineages based on the presence of the LDH-C*90 and LDH-C*100 alleles, respectively (Hamilton et al., 1989; García-Marín et al., 1999). Through mitochondrial analysis, five phylogenetic assemblages have been identified in Europe i.e. Adriatic, Danube, Mediterranean, marmoratus and Atlantic (Bernatchez et al., 1992).
It would appear that the geographical position of the Iberian Peninsula is crucial for brown trout distribution. Its rivers drain both into the Atlantic Ocean and the Mediterranean Sea, and some authors claim that the peninsula acted as a refuge during the last glaciations (García-Marín et al., 1999). Protein electrophoresis performed on Spanish brown trout populations suggests the existence of two distinct lineages associated with Atlantic and Mediterranean drainages (García-Marín & Pla, 1996). This has been confirmed recently using mitochondrial RFLPs by establishing five distinct groups, i.e. the Mediterranean, Andalusian, Atlantic, Duero and Cantabrian haplotypes (Machordom et al., 2000). However, this distribution pattern has not yet been related to that of other European areas.
The mitochondrial genome has been used widely to investigate phylogenetic relationships. As a consequence of its well-established organization, this genome has been sequenced in many organisms, including Salmo salar in the same genus as brown trout. With very few exceptions, the mitochondrial genome has a maternal mode of inheritance and has a mutation rate up to 10 times higher than nuclear single copy coding genes (Meyer, 1993; Avise, 2000). The mitochondrial control region, also called the displacement loop, or D-loop, is located between the genes coding for proline and phenylalanine tRNAs in vertebrates. It is responsible for the transcription of mitochondrial genes and contains the point of origin of heavy strand replication. Several regions of the mitochondrial D-loop show a high degree of variation between populations of the same species, but D-loop variability is not uniform and the region shows several well-conserved sequences among vertebrate species (Brown et al., 1986; Sbisà et al., 1997). The mitochondrial D-loop was selected as the molecular marker for the present study because it has been described as the most variable region of the mitochondrial molecule in terms of substitution rate and length variation (Bentzen et al., 1988; Hoelzel et al., 1991; Meyer, 1993; Cecconi et al., 1995). Furthermore, ample D-loop variability has been reported for brown trout throughout Europe (Bernatchez et al., 1992; Giuffra et al., 1994; Bernatchez & Osinov, 1995; Osinov & Bernatchez, 1996; Apostolidis et al., 1997; Weiss et al., 2000).
The aim of the present study was to analyse the variability of the S. trutta mitochondrial D-loop region and to perform a phylogenetic reconstruction of the populations of the Iberian Peninsula. Data from previous genetic studies on brown trout populations are reviewed in the light of present findings to infer phylogenetic relationships among European populations and complete the Palaearctic scenario of S. trutta evolution. Based on all of these data, a biogeographical hypothesis for the present brown trout distribution is presented.
Materials and methods
D-loop sequences and specimens
The complete DNA sequence of the D-loop region of 19 brown trout specimens was established (Table 1). Specimens represented all the haplotype groups present in the Iberian Peninsula as identified by RFLPs of a 2.7 kb mitochondrial DNA fragment, that comprised the genes NADH5 to Cyt-b (Machordom et al., 2000) plus one haplotype found thereafter (NA6). In most cases, each RFLP haplotype was represented by at least two specimens. Rare or unique RFLP haplotypes were each represented by one D-loop sequence (NA1, NA4, NA6, SA4 and ME4) (See Table 1). In addition to the 19 Iberian sequences analysed here, phylogenetic analysis of the European haplotypes was based on partial D-loop sequences retrieved from the GenBank database. These fragments corresponded to the Phe-tRNA and Pro-tRNA D-loop ends, which resulted in a 621-bp fragment of all the European brown trout haplotypes (GenBank accession numbers M97962-M97985) and one S. salar haplotype (GenBank accession numbers M97986, M97987). In all the complete and partial D-loop phylogenetic analyses, Oncorhynchus mykiss and S. salar (GenBank accession numbers L29771 and U12143, respectively) were used as outgroups.
Structural characterization of the S. trutta mitochondrial control region was achieved by aligning 18 complete sequences. These corresponded to the S. trutta consensus sequence of the present study, one human (J01415) and 16 teleost sequences (U12067, U12556, NC_002079, AF119321, AF140602, NC_001727, U12059, AF032381, AF06278, NC_001717, NC_002386, U12143, NC_000860). Complete teleost D-loop sequences corresponding to most teleost families were obtained from the GenBank database. Four Salmonidae sequences were similarly retrieved and aligned to identify hypervariable regions.
DNA extraction, amplification and sequencing
Total DNA extraction of frozen and alcohol-preserved muscle and liver tissues was conducted using standard phenol/chloroform protocols (Sambrook et al., 1989). Amplification of the brown trout mitochondrial control region was performed using the primers LN20 (5′-ACCACTAGCACCCAAAGCTA-3′) and HN20 (5′-GTGTTATGCTTTAGTTAAGC-3′) located in the proline and phenylalanine tRNA genes, respectively (Bernatchez & Danzmann, 1993); 50–150 ng of DNA were used to amplify the entire control region (∼1 kb). PCR amplification was performed according to standard procedures (1.25 U BIOTOOLS Taq-DNA polymerase, 2 mM MgCl2, 20 pmol of each primer and 250 μM dNTPs) for 40 cycles (92°C, 60 s; 50°C, 60 s; 72°C 90 s). Cycling was preceded by a 1-min denaturing step at 95°C and followed by a 10-min final extension at 72°C. Direct sequencing of both forward and reverse strands was performed for each amplified fragment in an ABI 377 automated system. Sequences were deposited in the GenBank database under the accession numbers AF253541–AF253559.
Mitochondrial DNA analysis
Sequences were aligned using CLUSTALX v1.5b software (Thompson et al., 1994). Conserved regions were visualized using the GENEDOC v2.3.0 program (Nicholas & Nicholas, 1997). Once the conserved regions had been identified, variable regions were realigned.
Phylogenetic hypotheses were established by minimum evolution (neighbour joining), maximum parsimony and maximum likelihood approaches. All phylogenetic analyses were performed using PAUP 4.0b3 (Swofford, 2000) including full heuristic searches by tree bisection reconnection (TBR) branch-swapping. Salmo salar and O. mykiss were run as multiple outgroups. Saturation tests were conducted to assess the reliability of the sequence analysis through all substitution types, and transitions and transversions alone. Likelihood ratio tests performed with MODELTEST 3.0 (Posada & Crandall, 1998), indicated model HKY85 with a gamma distribution and invariable sites as the model that best fitted the data. Nevertheless, the low number of nucleotide substitutions, and the topologies proposed by different variations of the HKY85 model, suggested the use of this model without the variables of rate variation across sites, proportion of invariable sites or gamma parameters, in the phylogeny estimation of brown trout for both distance and maximum likelihood analyses. This model is consistent with the brown trout D-loop sequence and nucleotide substitutions, i.e. sequences with nucleotide frequency bias and two substitution types (transitions vs. transversions). The transition:transversion ratio was estimated by maximum likelihood. Different weighting matrices (unweighted, weighting ts/tv, nucleotide frequencies and combined weighting) were used in the parsimony analyses, yielding similar results. Thus, the trees presented are those corresponding to the simplest, most parsimonious model. Incongruence–length difference tests (Farris et al., 1995) were performed to evaluate whether variable regions and distinct conserved regions resolved the same relationships. Three data partitions were tested (See Fig. 1): (i) The three domains of the mitochondrial control region; (ii) termination associated sequences (TAS), conserved sequence blocks (CSBs) and undefined sequences; and (iii) biologically meaningful elements vs. undefined sequences. Topologies were assessed by bootstrapping with 100 replicates in the distance and parsimony analyses, and by quartet puzzling steps (Strimmer & von Haeseler, 1996) with 1000 replicates in the maximum likelihood approach.

Mitochondrial D-loop structure of Salmo trutta. Black/grey/white boxes below the sequences indicate hypervariable/variable/conserved regions, respectively. The continuous line above the D-loop sequence highlights the TAS elements. The boxes within the sequence indicate CSBs.
Results
Characterization of the D-loop region
The 19 Iberian D-loop sequences ranged in size from 1025 to 1027 bp. Length variation was produced by single base-pair indels (n=6). Among the Iberian haplotypes, the maximum number of substitutions seen corresponded to 16 nucleotide changes between a South European Atlantic and an Andalusian haplotype (Table 2). Base composition in this region (63% A + T) corresponded to that of a typical A + T-rich region (Cecconi et al., 1995). Sequence change resulted predominantly from transitions rather than transversions, with a transition:transversion ratio of 2.39. Nucleotide substitutions occurred at 15 parsimony-informative characters and 16 autapomorphies. No repeated regions were detected in the S. trutta D-loop sequence.
Alignment of the S. trutta consensus sequence with teleost and human D-loop sequences revealed the general structure found in most vertebrates. The D-loop region was divided into three domains according to the amount of variability: (i) a left domain (Pro tRNA gene end); (ii) a right domain (Phe tRNA gene end); and (iii) a central domain (Fig. 1). TAS elements were found within the right domain and CSBs were found within central and left domains. The results of the incongruence–length difference tests of conserved features vs. variable regions were not significant for the three defined partitions (mitochondrial D-loop domains P=0.37, each separate element P=0.8, and biologically meaningful elements vs. undefined sequences P=1). Although the incongruence–length difference test seems to be conservative with the null hypothesis of homogeneity of partitions (Cunningham, 1997), the P-values obtained for the three defined partitions are far from the significance threshold of 0.05.
Phylogenetic relationships among brown trout
Pairwise comparison of the D-loop sequence of the specimens analysed served to identify two identical South European Atlantic specimens (SA1 and SA2). Three of the Mediterranean sequences (ME2, ME3 and ME4) were also identical, while one presented a unique mutation (ME1). All of the remaining sequences, i.e. the North Atlantic (NA1-NA6), Duero (DU1 and DU2), and Andalusian (AN1 and AN2) showed unique substitutions. Estimates of pairwise sequence divergence varied from 0 to 1.58% (mean ± SD, 0.74% ± 0.36).
Phylogenetic trees constructed through parsimony, minimum evolution and maximum likelihood analyses were of almost identical topology (Fig. 2). The 19 Iberian S. trutta sequences clustered into four monophyletic groups, i.e. Mediterranean, Andalusian, Atlantic and Duero. Character-based analysis resolved a total of 42 equally parsimonious trees (215 steps) in which all differences were shown within the Atlantic group and most within the South European Atlantic cluster.

Neighbour-joining tree based on the complete mitochondrial D-loop sequence of Iberian Peninsula Salmo trutta under the HKY85 model of evolution (transition:transversion ratio: 2.39). Numbers on branches represent the bootstrap values obtained for 100 replications corresponding to minimum evolution and maximum parsimony, and puzzling steps for 1000 replications corresponding to the maximum likelihood method. Support values under 50% are not represented. Abbreviations correspond to NA, North Atlantic; SA, South Atlantic; DU, Duero; ME, Mediterranean; AN, Andalusian.
Distance, parsimony and maximum likelihood analysis of the European D-loop sequences and the sequences presented in this study resolved the same six groups, although the relationships among them could not be fully resolved (Fig. 3). These groups were: (i) Mediterranean; (ii) Adriatic; (iii) Danube; (iv) Atlantic; (v) marmoratus; and (vi) Duero. The Mediterranean and Atlantic Iberian haplotypes were included in the Mediterranean and Atlantic European groupings, respectively. Andalusian haplotypes were included in the Adriatic group. Neither marmoratus nor Danube haplotypes were found within the Iberian Peninsula.

Maximum parsimony tree based on partial mitochondrial D-loop sequences of European Salmo trutta. Numbers on branches represent the bootstrap values obtained for 100 replications corresponding to maximum parsimony and minimum evolution, and puzzling steps for 1000 replications corresponding to the maximum likelihood method. Support values under 50% are not represented. Bold abbreviations as in Fig. 2. Plain text abbreviations represent the European mt D-loop haplotypes described; Bernatchez et al. (1992): Me, Mediterranean; At, Atlantic; Ad Adriatic; Da, Danube; Ma, marmoratus.
Brown trout biogeography
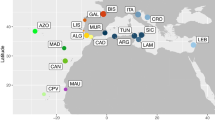
All of the phylogenetic groups showed a good correlation with geographical distribution (Fig. 4). North Atlantic haplotypes were found from the North Spanish coast to the White Sea. The Duero haplotypes showed a highly restricted distribution area confined to the Duero river, with few populations found in the Tajo. South European Atlantic haplotypes were identified in trout from the Atlantic-draining rivers of the Iberian Peninsula and also in the African specimen. This haplotype showed some overlap with the distribution pattern of the Duero haplotypes in the Duero basin, and with the North Atlantic haplotypes of populations from the Cantabrian Sea. Adriatic and Andalusian haplotypes, which constitute a phylogenetic cluster, were identified in specimens from two distinct areas, the Adriatic–Ionian region (Bernatchez et al., 1992; Giuffra et al., 1994; Apostolidis et al., 1997) and the southern Spanish basins of the present study. Mediterranean specimens showed a distribution range from the oriental basins of Spain to the Greek basins of the Adriatic Sea (Bernatchez et al., 1992; Giuffra et al., 1994; Apostolidis et al., 1997). Most of the area boundaries showed haplotype diversity forming contact zones between the different haplotype groups.

Model of the biogeographical areas across Europe as indicated by mitochondrial D-loop haplotypes. Dots indicate relative frequencies of each haplotype group: Atlantic (•), Duero ( ), Mediterranean (
), Mediterranean ( ), Adriatic - Andalusian (○), Salmo trutta marmoratus (
), Adriatic - Andalusian (○), Salmo trutta marmoratus ( ) and Danube (
) and Danube ( ).
).
Discussion
Phylogenetic relationships among brown trout
The four major haplotype groups found in the Iberian Peninsula (Atlantic, Duero, Mediterranean and Andalusian) were well supported. Although both North and South European Atlantic haplotypes showed low bootstrap values, the South European Atlantic grouping was resolved as monophyletic in maximum parsimony, minimum evolution and maximum likelihood analyses. This is consistent with previous findings derived from RFLP analysis of the 2.7-kb fragment of mitochondrial DNA (Machordom et al., 2000), in which both groups were also identified as monophyletic. The low sequence divergence within this cluster (mean 0.61%) and the high number of sequences analysed (11) would explain the low bootstrap values of the node separating North and South European Atlantic haplotypes. The topologies presented here show great similarity (Fig. 2) to the phylogeny of Iberian Peninsula populations based on RFLP analysis (Machordom et al., 2000), where identical groups were also defined. Nevertheless, some differences were found with respect to the internal nodes and relationships among groups. Although Mediterranean and Andalusian haplotypes appear as sister groups in both studies, according to RFLP analysis, the Duero cluster is basal to all the remaining haplotypes (Machordom et al., 2000). In the present analysis based on D-loop sequences, the Duero and Atlantic haplotypes appear as sister groups. The tree branches of these nodes are not well supported in either of these works. The short internodal distance in the RFLP analysis, observed together with the relatively low bootstrap support of the present tree (Fig. 2), indicates that the resolution of this cluster remains unclear. Independently of this cluster’s branching position, protein electrophoresis has clearly differentiated brown trout populations of Atlantic- and Mediterranean-draining rivers in the Iberian Peninsula (García-Marín & Pla, 1996), supporting its individuality.
At the European level, the topologies proposed here are consistent with the groups defined in other studies (Bernatchez et al., 1992; Giuffra et al., 1994; Apostolidis et al., 1997) but these studies were unable to resolve the relationships among groups. Andalusian and Adriatic haplotypes constituted an unexpected monophyletic group, which implies a wider distribution of this cluster in southern Europe than previously reported by other authors (i.e. Bernatchez et al., 1992; Giuffra et al., 1994; Apostolidis et al., 1997). Based on the discontinuous geographical distribution of this cluster, we renamed the Adriatic haplotype, Adriatic–Andalusian (see below for further discussion).
Brown trout biogeography
European brown trout populations exhibit biogeographical boundaries that serve to differentiate river basins and drainages as discrete units (Bernatchez et al., 1992; Giuffra et al., 1994; Apostolidis et al., 1997). These boundaries are also present in the Iberian Peninsula, with populations showing a high degree of haplotype diversity in relation to the different drainage systems, including Mediterranean, Atlantic and other endemic haplotypes (Machordom et al., 2000). Through the present D-loop analysis, congruent with the previously cited results, Atlantic haplotypes were segregated into two groups. The North Atlantic haplotypes were distributed from northern Spain to the White Sea, while the distribution of brown trout showing the South European Atlantic haplotype ran from northern Spain to the Atlantic coasts of Morocco. Duero haplotypes, found in populations from the Duero basin and some from the Tajo, represent an endemism of the Iberian Peninsula. Tajo populations are suspected to have acquired these haplotypes through river captures (Machordom et al., 2000). The presence of an Adriatic–Andalusian haplotype in specimens from southern Spain is remarkable, in particular when we consider that this haplotype has not been identified previously in populations of French or eastern Spanish rivers. We propose three hypotheses to explain the distribution of the Adriatic–Andalusian haplotype: (i) possible contact between Adriatic–Ionian and South Iberian populations through northwestern Africa (Morocco, Algeria and Tunisia); (ii) lineage sorting leading to the present haplotype distribution, with an ancestral polymorphism fixed at separate localities; and (iii) the extinction of Adriatic–Andalusian haplotypes in France and eastern Spain. Brown trout have been found in Algeria and the Mediterranean coasts of Morocco (Behnke, 1984). Hence, the study of these populations may provide evidence for the hypotheses presented here. The Adriatic Sea has been suggested as the origin for the Adriatic haplotypes that subsequently colonized Corsican and other Mediterranean basins via ancestral headwater connections (Bernatchez et al., 1992). However, based on its present distribution, it is impossible to establish an origin for the Adriatic–Andalusian haplotype.
Using the divergence rate of 0.8% per Myr corrected for Salmo trutta (Osinov & Bernatchez, 1996), the estimated divergence time between haplotypes corresponding to Atlantic and Mediterranean-draining rivers is in the range 1.2–1.6 Myr. This corresponds to the Pliocene-Pleistocene epoch. Divergence within the S. trutta complex was calculated to have occurred approximately 0.7–2 Myr ago (Ma) (Osinov & Bernatchez, 1996), an estimate consistent with the divergence time calculated from complete D-loop sequence alignments. Other approaches have indicated earlier divergence times for this species complex, i.e. 2.6–6.0 Ma (Apostolidis et al., 1997) or 0.4–6.3 Ma (Machordom et al., 2000). Thus, the different analytical methods appear to suggest two sets of divergence times between haplotypes. Estimates based on DNA sequences (Bernatchez et al., 1992; Giuffra et al., 1994; Osinov & Bernatchez, 1996) fall within 2% of the maximum divergence between Atlantic and Mediterranean drainage trout haplotypes, whereas estimates based on RFLPs (Machordom et al., 2000) or pooled DNA sequences and RFLPs (Apostolidis et al., 1997), unexpectedly indicate a value greater than 3% of the maximum divergence between the Atlantic and Mediterranean drainage population haplotypes. Sequence divergence estimates based on complete D-loop region sequences are in agreement with those derived from partial D-loop sequences (Bernatchez et al., 1992) and from pooled partial sequences corresponding to the D-loop, ATP synthase subunit VI and Cytochrome b (Giuffra et al., 1994). According to the present data, the Andalusian and Mediterranean haplotypes diverged 0.2–1.1 Ma, while the Duero and Atlantic haplotypes diverged 0.3–1.4 Ma. It is assumed that most significant genetic subdivisions within the brown trout complex are associated with major climatic change and basin isolation. The main Iberian hydrogeographic basins were established about 1.8–2.5 Ma (Calvo et al., 1993). This could explain present-day brown trout distribution in the Iberian Peninsula.
Glacial history seems to have played an important role in the diversity of North European Atlantic populations of S. trutta. Several hypotheses concerning the postglacial colonization of northern Europe have been proposed (Hamilton et al., 1989; García-Marín et al., 1999). Haplotype diversity shown by the southern populations of Europe and the low variability found in the North Atlantic populations suggest that glacial influence on the brown trout distribution was restricted to the north of the continent (Machordom et al., 2000; Weiss et al., 2000). The diversity of haplotypes found in the Iberian Atlantic rivers might represent an approximation to the haplotype diversity present in the northern Europe before the glaciations, when mitochondrial DNA variability would presumably have been higher. As temperatures decreased over the glacial period, the number of populations inhabiting the North decreased to extinction, erasing the haplotype variability of northern Europe; some of these populations survived in several glacial refuges (Hamilton et al., 1989; García-Marín et al., 1999). The Mediterranean populations were not affected by glaciations as much as those in northern Europe, thus maintaining their genetic variability.
In conclusion, the Iberian Peninsula maintains a haplotype variability that is not found throughout Europe. The present findings indicate that the Iberian Peninsula contains three of the five Salmo trutta groups previously described, and identifies a further different group, endemic to the Iberian Peninsula. The distribution of North Atlantic Mediterranean and Adriatic–Andalusian haplotypes is extended to the Iberian Peninsula, which could be considered a ‘hot-spot’ for brown trout diversity. Given that different levels of introgression with foreign populations have been recently detected (García-Marín et al., 1991; Morán et al., 1991; Martínez et al., 1993; Bouza et al., 1999; Machordom et al., 1999; Machordom et al., 2000), it is clear that conservation strategies such as avoiding foreign trout stock programmes are urgently needed to protect the endemic Duero and the mainly Iberian South European Atlantic haplotype. If restocking policies continue, trout stocks from all the biogeographical areas will be needed to maintain autochthonous genetic diversity in the Peninsula.
Accession codes
References
Apostolidis, A. P., Triantaphyllidis, C., Kouvatsi, A. and Economidis, P. S. (1997). Mitochondrial DNA sequence variation and phylogeography among Salmo trutta L. (Greek brown trout) populations. Mol Ecol, 6: 531–542.
Avise, J. C. (2000). Phylogeography: the History and Formation of Species, 1st edn. Harvard University Press, Cambridge, MA.
Behnke, R. J. (1972). The systematics of samonid fishes of recently glaciated lakes. J Fish Res Board Can, 29: 639–671.
Behnke, R. (1984). Salmonidae. In: Daget, J., Gosse, J.-P. and Thys, D. F. E. (eds) Check-List of the Freshwater Fishes of Africa (CLOFFA), Vol. 1, p. 125. Orstrom, Paris/Mrac, Tervuren.
Behnke, R. J. (1986). Brown trout. Trout, 27: 42–47.
Bentzen, P., Legget, W. C. and Brown, G. G. (1988). Length and restriction site heteroplasmy in the mitochondrial DNA of American shad (Alosa sapidissima). Genetics, 118: 509–518.
Bernatchez, L. and Danzmann, G. (1993). Congruence in control-region sequence and restriction-site variation in mitochondrial DNA of brook charr (Salvelinus fontinalis Mitchill). Mol Biol Evol, 10: 1002–1014.
Bernatchez, L. and Osinov, A. (1995). Genetic diversity of trout (genus Salmo) from its most eastern native range based on mitochondrial DNA and nuclear gene variation. Mol Ecol, 4: 285–297.
Bernatchez, L., Guyomard, R. and Bonhomme, F. (1992). DNA sequence variation of the mitochondrial control region among geographically and morphologically remote European brown trout Salmo trutta populations. Mol Ecol, 1: 161–173.
Bouza, C., Arias, J., Castro, J., Sánchez, L. and Martínez, P. (1999). Genetic structure of brown trout, Salmo trutta L., at the southern limit of the distribution range of the anadromous form. Mol Ecol, 8: 1991–2001.
Brown, G. G., Gadaleta, G., Pepe, G., Saccone, C. and Sbisà, E. (1986). Structural conservation and variation in the D-loop-containing region of vertebrate mitochondrial DNA. J Mol Evol, 192: 503–511.
Calvo, J. P., Daams, R., Morales, J., Lopez-Martinez, N., Agusti, J., Anadon, P. et al. (1993). Up-to-date Spanish continental Neogene synthesis and paleoclimatic interpretation. Rev Soc Geol España, 6: 29–40.
Cecconi, F., Giorgi, M. and Mariottini, P. (1995). Unique features in the mitochondrial D-loop region of the European seabass Dicentrarchus labrax. Gene, 160: 149–155.
Cunningham, C. W. (1997). Can three incongruence tests predict when data should be combined? Mol Biol Evol, 14: 733–740.
Farris, J. S., Källersjö, M., Kluge, A. G. and Bult, C. (1995). Testing significance of incongruence. Cladistics, 10: 315–319.
Ferguson, A. (1989). Genetic differences among brown trout, Salmo trutta, stocks and their importance for the conservation and management of the species. Freshwater Biol, 21: 35–46.
García-Marín, J. L. and Pla, C. (1996). Origins and relationships of native populations of Salmo trutta (brown trout) in Spain. Heredity, 77: 313–323.
García-Marín, J. L., Jorde, P. E., Ryman, N., Utter, F. and Pla, C. (1991). Management implications of genetic differentiation between native and hatchery populations of brown trout (Salmo trutta) in Spain. Aquaculture, 95: 235–249.
García-Marín, J. L., Utter, F. M. and Pla, C. (1999). Postglacial colonization of brown trout in Europe based on distribution of allozyme variants. Heredity, 82: 46–56.
Giuffra, E., Bernatchez, L. and Guyomard, R. (1994). Mitochondrial control region and protein coding genes sequence variation among phenotypic forms of brown trout Salmo trutta from northern Italy. Mol Ecol, 3: 161–171.
Hamilton, K. E., Ferguson, A., Taggart, J. B., Tómasson, T., Walker, A. and Fahy, E. (1989). Post-glacial colonization of brown trout, Salmo trutta L. Ldh-5 as a phylogeographic marker locus. J Fish Biol, 35: 651–664.
Hoelzel, A. R., Hancock, J. M. and Dover, G. A. (1991). Evolution of the Cetacean mitochondrial D-loop region. Mol Biol Evol, 8: 475–493.
Machordom, A., García-Marín, J. L., Sanz, N., Almodóvar, A. and Plá, C. (1999). Allozyme diversity in brown trout (Salmo trutta) from Central Spain: genetic consequences of restocking. Freshwater Biol, 41: 707–717.
Machordom, A., Suárez, J., Almodóvar, A. and Bautista, J. M. (2000). Mitochondrial haplotype variation and phylogeography of Iberian brown trout populations. Mol Ecol, 9: 1325–1338.
Martínez, P., Arias, J., Castro, J. and Sanchez, L. (1993). Differential stocking incidence in brown trout (Salmo trutta) populations from Northwestern Spain. Aquaculture, 114: 203–216.
Meyer, A. (1993). Evolution of mitochondrial DNA in fishes. In: Hochachka, P. W. and Mommsen, T. P. (eds) Biochemistry and Molecular Biology of Fishes, Vol. 2, pp. 1–38. Elsevier, Amsterdam.
Morán, P., Pendás, A. M., García-Vázquez, E. and Izquierdo, J. (1991). Failure of a stocking policy of hatchery reared brown trout, Salmo trutta L., in Asturias, Spain, detected using LDH-5* as a genetic marker. J Fish Biol, 39: 117–121.
Nicholas, K. B. and Nicholas, H. B. JR . (1997). GeneDoc: analysis and visualization of genetic variationhttp://www.cris.com/-ketchup/genedoc.shtml.
Osinov, A. G. and Bernatchez, L. (1996). ‘Atlantic’ and ‘Danubian’ phylogenetic groupings of brown trout Salmo trutta complex: genetic divergence, evolution, and conservation. J Ichthyol, 36: 723–746.
Posada, D. and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics, 14: 817–818.
Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular Cloning: a Laboratory Manual, 2nd edn., Cold Spring Harbor Laboratory Press, New York.
Sbisà, E., Tanzariello, F., Reyes, A., Pesole, G. and Saccone, C. (1997). Mammalian mitochondrial D-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene, 205: 125–140.
Strimmer, K. and von Haeseler, A. (1996). Quartet Puzzling: a Quartet Maximum-Likelihood Method for Reconstructing Tree Topologies. Mol Biol Evol, 13: 964–969.
Swofford, D. L. (2000). PAUP*: Phylogenetic analysis using parsimony (*And Other Methods). Sinauer, Sunderland, MA.
Thompson, J. D., Higgins, D. G. and Gibson, T. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res, 22: 4673–4680.
Weiss, S., Antunes, A., Schlötterer, C. and Alexandrino, P. (2000). Mitochondrial haplotype diversity among Portuguese brown trout Salmo trutta L. populations: relevance to the post-Pleistocene recolonization of northern Europe. Mol Ecol, 9: 691–698.
Acknowledgements
The authors wish to express their gratitude to S. Pérez-Benavente for her excellent technical assistance. They are also grateful to I. Doadrio, P. Martínez, P. Morán, J. Carmona and A.F. Alonso for providing supplementary brown trout material. DNA sequencing was performed at the automatic sequencing facility of the Universidad Complutense de Madrid. A. Burton reviewed the English. J.S. was supported by a predoctoral grant from the INIA (Instituto Nacional de Investigación y Tecnología Agraria y Agroalimentaria).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Suárez, J., Bautista, J., Almodóvar, A. et al. Evolution of the mitochondrial control region in Palaearctic brown trout (Salmo trutta) populations: the biogeographical role of the Iberian Peninsula. Heredity 87, 198–206 (2001). https://doi.org/10.1046/j.1365-2540.2001.00905.x
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1046/j.1365-2540.2001.00905.x
Keywords
This article is cited by
-
Applying genomic approaches to delineate conservation strategies using the freshwater mussel Margaritifera margaritifera in the Iberian Peninsula as a model
Scientific Reports (2022)
-
Biogeographic insights from a genomic survey of Salmo trouts from the Aralo-Caspian regions
Hydrobiologia (2022)
-
Genetic structure of important resident brown trout breeding lines in Poland
Journal of Applied Genetics (2020)
-
Development of a large SNPs resource and a low-density SNP array for brown trout (Salmo trutta) population genetics
BMC Genomics (2019)
-
Introgression despite protection: the case of native brown trout in Natura 2000 network in Italy
Conservation Genetics (2019)
