
Abstract
Aim:
Recent evidence suggests that aldo-keto reductase family 1 B10 (AKR1B10) may be a potential diagnostic or prognostic marker of human tumors, and that AKR1B10 inhibitors offer a promising choice for treatment of many types of human cancers. The aim of this study was to identify novel chemical scaffolds of AKR1B10 inhibitors using in silico approaches.
Methods:
The 3D QSAR pharmacophore models were generated using HypoGen. A validated pharmacophore model was selected for virtual screening of 4 chemical databases. The best mapped compounds were assessed for their drug-like properties. The binding orientations of the resulting compounds were predicted by molecular docking. Density functional theory calculations were carried out using B3LYP. The stability of the protein-ligand complexes and the final binding modes of the hit compounds were analyzed using 10 ns molecular dynamics (MD) simulations.
Results:
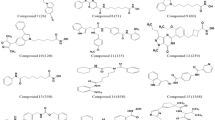
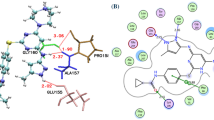
The best pharmacophore model (Hypo 1) showed the highest correlation coefficient (0.979), lowest total cost (102.89) and least RMSD value (0.59). Hypo 1 consisted of one hydrogen-bond acceptor, one hydrogen-bond donor, one ring aromatic and one hydrophobic feature. This model was validated by Fischer's randomization and 40 test set compounds. Virtual screening of chemical databases and the docking studies resulted in 30 representative compounds. Frontier orbital analysis confirmed that only 3 compounds had sufficiently low energy band gaps. MD simulations revealed the binding modes of the 3 hit compounds: all of them showed a large number of hydrogen bonds and hydrophobic interactions with the active site and specificity pocket residues of AKR1B10.
Conclusion:
Three compounds with new structural scaffolds have been identified, which have stronger binding affinities for AKR1B10 than known inhibitors.
Similar content being viewed by others
Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Gallego O, Ruiz FX, Ardevol A, Dominguez M, Alvarez R, de Lera AR, et al. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc Natl Acad Sci U S A 2007; 104: 20764–9.
Chung YT, Matkowskyj KA, Li H, Bai H, Zhang W, Tsao MS, et al. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod Pathol 2012; 25: 758–66.
Cao D, Liao DF . Author's reply to: AKR1B10 and its emerging role in tumor carcinogenesis and as a cancer biomarker. Int J Cancer 2013; 132: 496–7.
Kapoor S . AKR1B10 and its emerging role in tumor carcinogenesis and as a cancer biomarker. Int J Cancer 2013; 132: 495.
Fukumoto S, Yamauchi N, Moriguchi H, Hippo Y, Watanabe A, Shibahara J, et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers' non-small cell lung carcinomas. Clin Cancer Res 2005; 11: 1776–85.
Zhang W, Li H, Yang Y, Liao J, Yang GY . Knockdown or inhibition of aldo-keto reductase 1B10 inhibits pancreatic carcinoma growth via modulating Kras-E-cadherin pathway. Cancer Lett 2014; 355: 273–80.
Ruiz FX, Gallego O, Ardevol A, Moro A, Dominguez M, Alvarez S, et al. Aldo-keto reductases from the AKR1B subfamily: retinoid specificity and control of cellular retinoic acid levels. Chem Biol Interact 2009; 178: 171–7.
Balendiran GK, Martin HJ, El-Hawari Y, Maser E . Cancer biomarker AKR1B10 and carbonyl metabolism. Chem Biol Interact 2009; 178: 134–7.
Matsunaga T, Yamane Y, Iida K, Endo S, Banno Y, El-Kabbani O, et al. Involvement of the aldo-keto reductase, AKR1B10, in mitomycin-c resistance through reactive oxygen species-dependent mechanisms. Anticancer Drugs 2011; 22: 402–8.
Zhang L, Zhang H, Zhao Y, Li Z, Chen S, Zhai J, et al. Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: role of Trp112 (Trp111). FEBS Lett 2013; 587: 3681–6.
Wang C, Yan R, Luo D, Watabe K, Liao DF, Cao D . Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem 2009; 284: 26742–8.
Yan R, Zu X, Ma J, Liu Z, Adeyanju M, Cao D . Aldo-keto reductase family 1 B10 gene silencing results in growth inhibition of colorectal cancer cells: Implication for cancer intervention. Int J Cancer 2007; 121: 2301–6.
Matkowskyj KA, Bai H, Liao J, Zhang W, Li H, Rao S, et al. Aldoketoreductase family 1B10 (AKR1B10) as a biomarker to distinguish hepatocellular carcinoma from benign liver lesions. Hum Pathol 2014; 45: 834–43.
Soda M, Hu D, Endo S, Takemura M, Li J, Wada R, et al. Design, synthesis and evaluation of caffeic acid phenethyl ester-based inhibitors targeting a selectivity pocket in the active site of human aldo-keto reductase 1B10. Eur J Med Chem 2012; 48: 321–9.
Takemura M, Endo S, Matsunaga T, Soda M, Zhao HT, El-Kabbani O, et al. Selective inhibition of the tumor marker aldo-keto reductase family member 1B10 by oleanolic acid. J Nat Prod 2011; 74: 1201–6.
Endo S, Matsunaga T, Kuwata K, Zhao HT, El-Kabbani O, Kitade Y, et al. Chromene-3-carboxamide derivatives discovered from virtual screening as potent inhibitors of the tumour maker, AKR1B10. Bioorg Med Chem 2010; 18: 2485–90.
Endo S, Hu D, Suyama M, Matsunaga T, Sugimoto K, Matsuya Y, et al. Synthesis and structure-activity relationship of 2-phenyliminochromene derivatives as inhibitors for aldo-keto reductase (AKR) 1B10. Bioorg Med Chem 2013; 21: 6378–84.
Niu MM, Qin JY, Tian CP, Yan XF, Dong FG, Cheng ZQ, et al. Tubulin inhibitors: pharmacophore modeling, virtual screening and molecular docking. Acta Pharmacol Sin 2014; 35: 967–79.
Sakkiah S, Lee KW . Pharmacophore-based virtual screening and density functional theory approach to identifying novel butyrylcholinesterase inhibitors. Acta Pharmacol Sin 2012; 33: 964–78.
Li H, Sutter J, Hoffmann R . HypoGen: An automated system for generating predictive 3D pharmacophore models. In: Güner OF, editor. Pharmacophore Perception, Development, and use in Drug Design. California: International University Line; 2000. p 171–189.
Sakkiah S, Thangapandian S, John S, Kwon YJ, Lee KW . 3D QSAR pharmacophore based virtual screening and molecular docking for identification of potential HSP90 inhibitors. Eur J Med Chem 2010; 45: 2132–40.
Debnath AK . Pharmacophore mapping of a series of 2,4-diamino-5-deazapteridine inhibitors of mycobacterium avium complex dihydrofolate reductase. J Med Chem 2002; 45: 41–53.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ . Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001; 46: 3–26.
Yang SY . Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 2010; 15: 444–50.
Jones G, Willett P, Glen RC, Leach AR, Taylor R . Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997; 267: 727–48.
Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD . Improved protein-ligand docking using GOLD. Proteins 2003; 52: 609–23.
Tsuneda T . Orbital energy. In: Tsuneda T, editor. Density functional theory in quantum chemistry. Japan: Springer; 2014. p 161–188
Eroglu E, Turkmen H . A DFT-based quantum theoretic QSAR study of aromatic and heterocyclic sulfonamides as carbonic anhydrase inhibitors against isozyme, CA-II. J Mol Graph Model 2007; 26: 701–8.
Ai C, Li Y, Wang Y, Li W, Dong P, Ge G, et al. Investigation of binding features: effects on the interaction between CYP2A6 and inhibitors. J Comput Chem 2010; 31: 1822–31.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ . GROMACS: fast, flexible, and free. J Comput Chem 2005; 26: 1701–18.
Zoete V, Cuendet MA, Grosdidier A, Michielin O . SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 2011; 32: 2359–68.
Bussi G, Donadio D, Parrinello M . Canonical sampling through velocity rescaling. J Chem Phys 2007; 126: 014101.
Parrinello M, Rahman A . Polymorphic transitions in single crystals. A new molecular dynamics method. J Appl Phys 1981; 52: 7182.
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM . LINCS: a linear constraint solver for molecular simulations. J Comput Chem 1997; 18: 1463–72.
Darden T, York D, Pedersen L . Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys 1993; 98: 10089.
Son M, Baek A, Sakkiah S, Park C, John S, Lee KW . Exploration of virtual candidates for human HMG-CoA reductase inhibitors using pharmacophore modeling and molecular dynamics simulations. PLoS One 2013; 8: e83496.
Humphrey W, Dalke A, Schulten K . VMD: visual molecular dynamics. J Mol Graph 1996; 14: 33–8.
Hou T, Wang J, Li Y, Wang W . Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011; 51: 69–82.
Spiliotopoulos D, Spitaleri A, Musco G . Exploring PHD fingers and H3K4me0 interactions with molecular dynamics simulations and binding free energy calculations: AIRE-PHD1, a comparative study. PLoS One 2012; 7: e46902.
Sonawane KD, Barage SH . Structural analysis of membrane-bound hECE-1 dimer using molecular modeling techniques: insights into conformational changes and Aβ1–42 peptide binding. Amino Acids 2014. doi: 10.1007/s00726-014-1887-8 Epub 2014 Dec 16.
Genheden S, Ryde U . How to obtain statistically converged MM/GBSA results. J Comput Chem 2010; 31: 837–46.
Vorontsov II, Miyashita O . Crystal molecular dynamics simulations to speed up MM/PB(GB)SA evaluation of binding free energies of di-mannose deoxy analogs with P51G-m4-Cyanovirin-N. J Comput Chem 2011; 32: 1043–53.
Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP . Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des 1997; 11: 425–45.
Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, et al. Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem 2007; 50: 726–41.
Wang Y, Suzek T, Zhang J, Wang J, He S, Cheng T, et al. PubChem BioAssay: 2014 update. Nucleic Acids Res 2014; 42 (Database issue): D1075–82.
Bonnet P, Bryce RA . Molecular dynamics and free energy analysis of neuraminidase–ligand interactions. Protein Sci 2004; 13: 946–57.
Donini OA, Kollman PA . Calculation and prediction of binding free energies for the matrix metalloproteinases. J Med Chem 2000; 43: 4180–8.
Wright DW, Hall BA, Kenway OA, Jha S, Coveney PV . Computing clini-cally relevant binding free energies of HIV-1 protease inhibitors. J Chem Theory Comput 2014; 10:1228–41.
Gilson MK, Zhou HX . Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct 2007; 36: 21–42.
Acknowledgements
This research was supported by the Basic Science Research Program (2012R1A1A4A01013657) and Management of Climate Change Program (2010-0029084) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education of Republic of Korea. This work was also supported by the Next-Generation BioGreen 21 Program (PJ009486) from the Rural Development Administration (RDA) of Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information is available at http://www.chinaphar.com/
Supplementary information
Supplementary Information, Table 1
Chemical structures of test set compounds with their respective IC50 values. (DOC 429 kb)
PowerPoint slides
Rights and permissions
About this article

Cite this article
Kumar, R., Son, M., Bavi, R. et al. Novel chemical scaffolds of the tumor marker AKR1B10 inhibitors discovered by 3D QSAR pharmacophore modeling. Acta Pharmacol Sin 36, 998–1012 (2015). https://doi.org/10.1038/aps.2015.17
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/aps.2015.17
Keywords
This article is cited by
-
Identification of novel leads as potent inhibitors of HDAC3 using ligand-based pharmacophore modeling and MD simulation
Scientific Reports (2022)
-
New compounds identified through in silico approaches reduce the α-synuclein expression by inhibiting prolyl oligopeptidase in vitro
Scientific Reports (2017)
-
Identification of potential type 4 cAMP phosphodiesterase inhibitors via 3D pharmacophore modeling, virtual screening, DFT and structural bioisostere design
Medicinal Chemistry Research (2017)

{kind=link}
{kind=link}