
Abstract
Background:
NGR–hTNF exploits the peptide asparagine–glycine–arginine (NGR) for selectively targeting tumour necrosis factor (TNF) to CD13-overexpressing tumour vessels. Maximum-tolerated dose (MTD) of NGR–hTNF was previously established at 45 μg m−2 as 1-h infusion, with dose-limiting toxicity being grade 3 infusion-related reactions. We explored further dose escalation by slowing infusion rate (2-h) and using premedication (paracetamol).
Methods:
Four patients entered each of 12 dose levels (n=48; 60–325 μg m−2). Pharmacokinetics, soluble TNF receptors (sTNF-R1/sTNF-R2), and volume transfer constant (Ktrans) by dynamic imaging (dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI)) were assessed pre- and post-treatment.
Results:
Common related toxicity included grade 1/2 chills (58%). Maximum-tolerated dose was not reached. Both Cmax (P<0.0001) and area under the plasma concentration–time curve (P=0.0001) increased proportionally with dose. Post-treatment levels of sTNF-R2 peaked significantly higher than sTNF-R1 (P<0.0001). Changes in sTNF-Rs, however, did not differ across dose levels, suggesting a plateau effect in shedding kinetics. As best response, 12/41 evaluable patients (29%) had stable disease. By DCE-MRI, 28/37 assessed patients (76%) had reduced post-treatment Ktrans values (P<0.0001), which inversely correlated with NGR–hTNF Cmax (P=0.03) and baseline Ktrans values (P<0.0001). Lower sTNF-R2 levels and greater Ktrans decreases after first cycle were associated with improved survival.
Conclusion:
asparagine–glycine–arginine–hTNF can be safely escalated at doses higher than MTD and induces low receptors shedding and early antivascular effects.
Similar content being viewed by others

Main
Tumour necrosis factor (TNF)-alpha is a powerful antitumour cytokine originally identified (Carswell et al, 1975) for its ability to induce in experimental cancers massive haemorrhagic necrosis, which is mainly mediated by apoptosis of cancer endothelial cells. The apoptotic pathway is triggered by TNF binding with the receptor TNF-R1. Conversely, the receptor TNF-R2 lacks a death domain and likely acts as a modulator of TNF-R1 actions. Both receptors are also shed as soluble proteins, thus competing with cell-surface receptors for free ligand (Balkwill, 2009). Early clinical trials with TNF given systemically were, however, associated to severe toxicities, with maximum-tolerated dose (MTD) being significantly lower than estimated effective dose (Lejeune et al, 2006). Lately, through isolated limb perfusion it was possible to deliver loco-regionally doses of TNF 10-fold higher than MTD. Of note, TNF in combination with chemotherapy induced high response rates in several tumours, with an acceptable toxicity profile (Fraker et al, 1995).
In order to increase the TNF therapeutic index, a ligand-directed vascular-targeting approach was exploited. For this purpose, NGR–hTNF was prepared by conjugating the N-terminus of TNF with the C-terminus of the tumour-homing peptide NGR (asparagine–glycine–arginine) that selectively recognises an aminopeptidase N (CD13) isoform overexpressed by angiogenic tumour vessels (Arap et al, 1998; Curnis et al, 2000). Recently, CD13 was found to be crucial for the pathological development of newly formed blood vessels from preexisting ones in CD13-null mice (Rangel et al, 2007). Compared with untargeted TNF, NGR–TNF induced a significantly increased antitumour activity in preclinical models and, more interestingly, displayed a biphasic (U-shaped) dose–response curve with activity noted either at very low (⩽0.1 ng) or at high doses (>1000 ng) (Curnis et al, 2002).
A previously reported phase I study (van Laarhoven et al, 2010) testing doses ranging from 0.2 to 60 μg m−2 established the MTD of NGR–hTNF at 45 μg m−2 given intravenously as 1-h infusion once every 3 weeks, with dose-limiting toxicities (DLTs) encountered at the dose of 60 μg m−2 being grade 3 infusion-related reactions (IRRs). Common related toxicities were transient grade 1 to 2 chills (58% of patients) and fever (56%). Shedding of soluble TNF receptors was not detected at the lowest doses tested (⩽0.8 μg m−2), while thereafter increased proportionally with dose up to a plateau observed at ⩾25 μg m−2. No objective responses were detected, but of six patients with durable stable diseases, five were treated with low doses.
An additional trial exploring the low-dose range from 0.2 to 1.6 μg m−2 selected 0.8 μg m−2 as optimal biological low dose, based on dynamic imaging changes and sTNF-Rs shedding kinetics (Gregorc et al, 2010). In this study, the antivascular effects of NGR–hTNF were confirmed by dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), with 75% of patients having decreased values of the volume transfer constant, Ktrans. Considering the preclinical activity noted only at very high doses and the temporal relationship between DLTs onset and drug dosing, we decided to explore higher doses of NGR–hTNF using a more protracted infusion length of 2 h and a mild premedication with paracetamol.
The present study primarily aimed at determining the optimal biological high dose of NGR–hTNF, given at doses higher than the previously established MTD, by evaluating both safety, in terms of MTD, and antivascular effects, in terms of changes in dynamic imaging. Secondary aims included pharmacokinetics, pharmacodynamics, and preliminary activity.
Methods
Patients eligibility
Patients 18 years of age or older with refractory solid tumours and radiological disease progression after last treatment regimen were enroled. Additional eligibility requirements were: ECOG performance status (PS) of 0 to 1; absolute neutrophil count >1.5 × 109 l−1; platelet count >100 × 109 l−1; total bilirubin <1.5 × upper limit of normal (ULN); aspartate and alanine aminotransferase <2.5 × ULN in the absence of liver metastasis or <5 × ULN in the presence of liver metastasis; and serum creatinine <1.5 × ULN. Patients with significant cardiac, infectious, or peripheral vascular diseases were excluded, as well as patients completing systemic therapy within 4 weeks or having surgery within 2 weeks before treatment start. The Institutional Review Board approved this study, which was conducted in accordance with the Declaration of Helsinki. Signed informed consent was obtained from all patients before trial participation. Trial number registration at clinicaltrials.gov is NCT00878111.
Study design
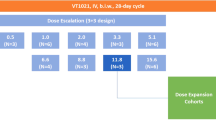
This was a single-centre, dose-defining study with a minimum of four patients who were administered each of 12 dose levels (DLs: 60–80–100–125–150–175–200–225–250–275–300–325 μg m−2). The starting dose (60 μg m−2) was 33% higher than the previously selected MTD and recommended phase II dose (45 μg m−2) (van Laarhoven et al, 2010). To have a more robust toxicity assessment at each DL, four instead of three patients were enroled and a 25-μg flat dose increase was cautiously used from the third level. Patients were treated at their assigned dose of NGR–hTNF that was given as 2 hours infusion every 3 weeks until progression, unacceptable toxicity, or patient request. Premedication with paracetamol (1000 mg per os or intravenously) was mandated 30 to 60 min before each infusion. No dose modification was allowed. For retreatment on next cycle, all related toxicities should be recovered to grade 1 or less. If a patient was unable to meet retreatment criteria, NGR–hTNF was delayed for 1 week for up to 3 weeks. In the presence of infusion-related symptoms of grade ⩾2, the rate was slowed/interrupted and patients monitored/treated until complete resolution.
Adverse events (AEs) were graded according to Common Terminology Criteria for Adverse Events (version 3.0). Dose-limiting toxicities applicable to the study were defined as any grade 3 or 4 toxicity clearly related to NGR–hTNF. Exceptions were considered nausea, vomiting, chills, and fever that could be rapidly controlled with appropriate treatments. Four patients were to be enroled to each DL. After enrolment of the first two patients, new patient inclusion was blocked until the safety of the first two patients to the first cycle was fully documented in order to verify that no DLT had occurred. Then, subsequent two patients were allowed for registration at that particular DL. If one or less of the four patients experienced a DLT during the first cycle, an additional four patients were entered at the next higher DL with dose escalation continuing until DLT was observed. If two or more of the four patients experienced DLT during the first cycle, the prior lower DL would be declared as MTD.
Pharmacokinetic and pharmacodynamic assessment
Intensive pharmacokinetic (PK) blood sampling was performed on day 1 of the first 3 cycles with samples drawn at baseline (before NGR–hTNF dosing) and on treatment (20–60–90–120–180–240 min after each dosing). The plasma levels of NGR–hTNF and soluble TNF receptors (sTNF-R1 and sTNF-R2) were computed using an enzyme-linked immunosorbent assay. Maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) of NGR–hTNF were estimated from plasma concentration–time data using standard noncompartmental methods. Pharmacodynamic variables determined for sTNF-Rs were Emax (maximum plasma concentration) and AUC. Because of the background levels of TNF and sTNF-Rs generally detectable in cancer patients, concentration–time profiles for each patient were baseline-normalised by subtracting the time-zero value to all other time-point values.
Dynamic contrast-enhanced magnetic resonance imaging
Magnetic resonance imaging studies were performed on a 1.5T MR scanner and images were analysed as previously described (d’Arcy et al, 2006). Dynamic contrast-enhanced magnetic resonance imaging scanning were done at baseline (48 h before first dose) and 2 h after administration of first and subsequent cycles. This 2-h interval was chosen because maximal synergism of NGR–hTNF with cytotoxic agents was observed with a 2-h delay between NGR–hTNF and chemotherapy administration (Sacchi et al, 2006), thus suggesting a maximum drug effect on the vasculature at that time point. The gadopentetate dimeglumine concentration curve was fitted with the standardised Tofts PK model (Tofts et al, 1999), and maps of Ktrans (volume transfer constant from plasma to extravascular extracellular leakage space, EES) were estimated. Ktrans depends on balance between capillary permeability and blood flow in the tissue of interest. In high-permeability situations, where tracer flux is flow limited, Ktrans is equal to the blood plasma flow per unit volume of tissue. In low-permeability situations, where tracer flux is permeability limited, Ktrans is equal to the permeability surface area product between blood plasma and EES, per unit volume of tissue. Thus, reductions in Ktrans would mean either reductions in tumour vascularity (i.e., the amount of capillaries) or reductions in tumour permeability, or both situations. The initial area under concentration agent–time curve at 90 s (IAUGC) as a semiquantitative index was also obtained. Regions of interest were traced twice by the same expert radiologist in areas of tumour tissue with intense enhancement. Given that a repeatability coefficient was not formally defined in the present study, reductions from baseline in Ktrans or IAUGC >50% were considered a significant antivascular effect (Jackson et al, 2007; O’Connor et al, 2012).
Statistical analysis
Descriptive statistics were provided using medians with 95% confidence interval (CI) or means with s.d. for continuous variables and proportions for categorical variables. Continuous data were compared by the nonparametric Mann–Whitney test, whereas the degree of association between continuous variables was quantified by the Spearman rank correlation coefficient. A Kruskal–Wallis analysis of variance by ranks was used to assess dose proportionality and compare differences in baseline-normalised sTNF-Rs as a function of dose. For DCE-MRI parameters, absolute values and relative percentage changes from baseline were reported, and to determine if the distribution of these changes between pairwise time points differed significantly from zero, the Wilcoxon test was used. Baseline-normalised AUC and Cmax were used as representative parameters of drug exposure and were correlated with changes in dynamic imaging parameters. Measurable target lesions were radiologically assessed using response evaluation criteria in solid tumours (RECIST version 1.0), or mesothelioma-modified RECIST criteria (Byrne and Nowak, 2004), while Kaplan–Meyer method and log-rank test were used for comparisons of time-to-event data between patient subgroups.
Results
Patients
In all, 48 patients (37 men and 11 women) with a median age of 61 years (range, 23–76 years) and a PS of 0 (n=21) or 1 (n=27) were enroled. Most of the patients were affected by colon cancer (n=28), followed by mesothelioma (n=8), liver cancer (n=5), soft-tissue sarcoma (n=4), gastric cancer (n=2), and pancreatic neuroendocrine tumour (PNET; n=1). All patients were heavily pretreated with a median of three systemic regimens (range, one to seven).
Safety
A total of 117 cycles (median, two; range, one to six cycles) were delivered. In all the 48 enroled patients, treatment discontinuations resulted from symptomatic deterioration (n=7) or radiological tumour progression (n=41).
The MTD was not reached and no DLTs were observed. Overall, NGR–hTNF was well tolerated without apparent differences in either frequency or intensity of AEs by dose. Most common AEs over all cycles, regardless of drug relationship, were pyrexia and chills (Table 1), with grade 3 and 4 AEs reported by 12 (25%) and 5 (10%) patients, respectively. No patient experienced grade 3–4 drug-related AEs, while grade 1–2 IRRs included chills (58%), vomiting (44%), and hypotension (25%). During the first cycle, chills was noted in 24 patients (50%).
Pharmacokinetics
Overall, 46 patients had PK studies completed. After the first cycle, the mean (±s.d.) Cmax ranged from 0.1 (±0.1) at the dose of 60 μg m−2 to 44.2 (±6.4) ng ml−1 at the dose of 325 μg m−2, whereas the corresponding AUC values ranged from 14 (±6) to 4966 (±548) ng h−1 ml−1, respectively. Concentration–time profiles after the first three cycles by dose are depicted in Figure 1A. Both Cmax (P<0.0001) and AUC (P=0.0001; Figure 1B) increased proportionally with dose.

Pharmacokinetic results over the first three treatment cycles by dose levels (n=46). (A) Baseline-normalised concentration–time profiles individual Cmax and (B) individual AUC of NGR–hTNF. Four patients were analysed at each DL with the exception of 175 (n=3) and 300 μg m−2 (n=3).
Pharmacodynamics
Baseline and post-treatment plasma levels of sTNF-Rs were monitored in 46 patients. After the first cycle, the concentrations of sTNF-R1 and sTNF-R2 were significantly increased (P<0.0001 for both). Baseline-normalised Emax of sTNF-R2 (median, 9.6 ng ml−1; 95% CI, 8.7–10.2) peaked significantly higher than sTNF-R1 (median, 4.9 ng ml−1; 95% CI, 4.6–5.4; P<0.0001). However, the concentration–time profiles of sTNF-R1 and sTNF-R2 did not significantly differ across DLs, suggesting a plateau effect in shedding kinetics.
The antivascular effects of NGR–hTNF were evaluated in 37 patients who had at least one post-baseline DCE-MRI assessment. As shown in Table 2, both Ktrans and IAUGC were markedly reduced after first cycle with an apparent dose-dependent effect, as the changes in Ktrans correlated with both Cmax (r=−0.35; P=0.03) and AUC (r=−0.31; P=0.06) of NGR–hTNF. Overall, 28 patients (76%; 95% CI, 59–88%) had reduced values of Ktrans and IAUGC at some point on treatment, and 16 patients (43%; 95% CI, 27–60%) experienced decreases in Ktrans from baseline >50% (Figure 2). In particular, patients receiving DLs ⩾200 μg m−2 had significantly greater reductions in Ktrans than patients receiving DLs <200 μg m−2 (median, −52% and −24%, respectively; P<0.0001) (Figure 3A). Moreover, for higher baseline Ktrans values, greater decreases were noted. Indeed, the absolute changes in Ktrans were inversely correlated with pre-treatment values (r=−0.58; P=0.0002), particularly in the subset of 28 patients who had decreased values (r=−0.85; P<0.0001) (Figure 3B). There was no correlation between pre-treatment tumour size and either baseline or post-treatment Ktrans values. Decreases in Ktrans were reported in 16/22 patients with colon cancer, in 5/6 with mesothelioma, in 2/4 with sarcomas, in 3/3 with liver cancer, and in one patient each with gastric cancer and PNET.

Dynamic contrast-enhanced magnetic resonance imaging assessment (n=37). Maximal changes from baseline in Ktrans by dose levels. The dotted line denotes the threshold reduction of 50% in Ktrans, considered a significant antivascular effect.

Dynamic contrast-enhanced magnetic resonance imaging assessment (n=37). (A) Relationships between percentage changes in Ktrans after first cycle and dose levels grouped between lower (from 80 to 175 μg m−2) and higher (from 200 to 325 μg m−2) doses. The horizontal solid lines denote median values. (B) Relationships between baseline Ktrans values and absolute changes in Ktrans.
Antitumour activity
Overall, 12 (29%; 95% CI, 16–45%) of 41 patients evaluable for best response by RECIST criteria had stable disease. There were no significant relationships between changes in DCE-MRI parameters and RECIST-defined tumour response, though patients with stable disease tended to have greater reductions in Ktrans after first cycle than patients with early progression (median, −48 and −28%, respectively). Median progression-free survival (PFS) was 1.7 months (95% CI, 1.5–1.9 months) and 1-year overall survival (OS) rate was 34% (95% CI, 17–51%). The subset of patients with stable disease (including eight with colorectal cancer, two with mesothelioma, and one each with gastric cancer and PNET) had median PFS of 2.9 months and median OS of 13.0 months. Patients who reported chills during the first infusion had significantly improved PFS (hazard ratio=0.50; P=0.009) compared with patients who did not. Moreover, lower levels of sTNF-R2 and greater decreases in Ktrans were detected after the first cycle in patients who subsequently experienced prolonged survival time. In particular, median OS was 13.0 months in patients with sTNF-R2 levels <9.6 ng ml−1 (i.e., the median distribution value) and 3.9 months in patients with levels ⩾9.6 ng ml−1 (P=0.004), whereas median OS was 14.5 months in patients with decreases in Ktrans >50% and 4.7 months in patients with decreases ⩽50% (P=0.05).
Discussion
Drug doses are established in phase I studies, but infusion length or administration frequency are commonly determined empirically based on available preclinical data. Then, subsequent observations can either reinforce or modify these decisions. However, often dosing schedules are based on other considerations, such as volume necessary for administration or, most importantly, safety.
The MTD of NGR–hTNF was established at 45 μg m−2 as 1-h intravenous infusion in a previous study testing a dose interval from 0.2 to 60 μg m−2, with DLTs consisting of grade 3 IRRs (van Laarhoven et al, 2010). In that study, the infusion length was not further prolonged because it was assumed that, given a dose-proportional increase of NGR–hTNF exposure and a plateau of sTNF-Rs shedding, the excess of free drug unbound to circulating receptors at higher doses would be responsible for DLTs. Therefore, further prolongation of the infusion time was not expected to alter toxicity.
Though severe reactions are rare, mild-to-moderate IRRs occur frequently with many compounds and tend to resolve after brief infusion interruption and supportive care administration (Lenz, 2007). Furthermore, most patients experiencing mild-to-moderate IRRs during first exposure tend to tolerate drug rechallenge with slower infusion rate and premedication.
Therefore, in the present study a more prolonged infusion time empirically set at 2 h was explored, so that if IRRs were detected, the infusion could be interrupted with a relatively small amount of drug having been delivered. Moreover, considering that the vast majority of these IRRs consisted of chills, a mild premedication with paracetamol was mandated starting from first cycle.
The present study has clearly demonstrated that NGR–hTNF can be safely escalated at doses up to seven times higher than the previous MTD by slowing the infusion rate at 2 h and using a mild premedication with paracetamol. The use of high doses did not change the toxicity pattern reported for single-agent NGR–hTNF, mainly consisting of mild-to-moderate and dose-unrelated chills. Similarly, the cardiovascular toxicities described for vascular-targeting agents (including acute coronary and thrombophlebitic syndromes; alterations in blood pressure, heart rate, and ventricular conduction) (van Heeckeren et al, 2006) did not appear in this study, likely because of targeted delivery of this compound.
As expected using a targeted agent and despite the wide dose interval tested, the present trial did not formally define a new MTD. However, the present study primarily aimed at determining the optimal biological high dose of NGR–hTNF by evaluating both safety and antivascular effects by dynamic imaging. Furthermore, it is now widely agreed that the toxicity spectrum of targeted agents differs profoundly from the one of cytotoxic agents and, given their cytostatic nature and lack of dose–effect relationship, increasing drug dose to MTD may be unnecessary for drug effect (Ellis and Hicklin, 2008).
Notwithstanding the fact that the selection of an optimal biological dose is highly challenging, it is worthy to note that the low dose of 0.8 μg m−2 was previously selected based on more pronounced antivascular effects and no shedding of soluble TNF receptors noted at this dose level (Gregorc et al, 2010). These circulating receptors might compete with the cell-surface receptors for free TNF, thus blocking its bioavailability and activity, with the amount and speed of this shedding being linearly correlated with serum TNF level (Aderka et al, 1998). The plateau in sTNF-Rs shedding kinetics observed previously (van Laarhoven et al, 2010) and confirmed also in the present study is consistent with a biphasic dose–response curve and suggests that high doses of NGR–hTNF can overcome this counterregulatory mechanism.
DCE-MRI scans were done at baseline and 2 h after NGR–hTNF dosing to measure the biological effects on tumour vascularity. Shortly after a single dose, NGR–hTNF markedly decreased Ktrans, and doses higher than 200 μg m−2 appeared to be the most effective, with median reductions of >50%. It is important to emphasise that Ktrans is a mixed measure of tumour vascularity and permeability. Thus, Ktrans measurements could be influenced by several factors, including tumour types and analysis models. In the present study, there was no indication that any particular tumour type was overrepresented among tumours with either more dramatic decreases or higher baseline Ktrans values. Interestingly, a correlation between baseline values and post-treatment changes in Ktrans was noted, thus suggesting increased antivascular effects in tumours with extensive abnormal vasculature.
The fact that these decreases in tumour permeability and blood flow occur so rapidly can be interpreted either as normalisation of tumour vasculature owing to effects on integrity (permeability) and function (vessel perfusion) of vascular bed, or vascular damage. Both mechanisms are expected, however, to improve the synergy with chemotherapy by increasing the intratumoural chemotherapy uptake or sensitising the tumour vascular compartment to cytotoxic effects. Therefore, both mode of action of NGR–hTNF and nonoverlapping toxicity profile with cytotoxic agents could facilitate the combination with chemotherapy. Consistently, NGR–hTNF has been reported to synergise with several chemotherapeutic agents (Sacchi et al, 2006).
Even though a short median PFS of 1.7 months was registered for the overall study population, this figure seems to be substantially in line with median PFS of 2.5 months reported in literature for heavily pretreated patients enroled in phase I trials (Arkenau et al, 2008). Interestingly, the early post-treatment findings that the lower the Ktrans and sTNF-Rs values, the higher the patient survival rates, deserve further investigations in larger patient population to clarify their role as suitable tools for either prognostication or prediction.
In conclusion, this study has shown that NGR–hTNF can be safely given at doses higher than the previous MTD using a mild premedication and a longer infusion time. The observed tolerability profile and biological effects warrant further clinical testing of doses higher than 200 μg m−2 of NGR–hTNF combined with chemotherapy and compared with a low dose of 0.8 μg m−2.
Change history
15 January 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aderka D, Sorkine P, Abu-Abid S, Lev D, Setton A, Cope AP, Wallach D, Klausner J (1998) Shedding kinetics of soluble tumour necrosis factor (TNF) receptors after systemic TNF leaking during isolated limb perfusion. J Clin Invest 101: 650–659
Arap W, Pasqualini R, Ruoslahti E (1998) Cancer treatment by targeted drug delivery to tumour vasculature in a mouse model. Science 279: 377–380
Arkenau HT, Olmos D, Ang JE, de Bono J, Judson I, Kaye S (2008) Clinical outcome and prognostic factors for patients treated within the context of a phase I study: the Royal Marsden Hospital experience. Br J Cancer 98: 1029–1033
Balkwill F (2009) Tumour necrosis factor and cancer. Nat Rev Cancer 9: 361–371
Byrne MJ, Nowak AK (2004) Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Ann Oncol 15: 257–260
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumours. Proc Natl Acad Sci USA 72: 3666–3670
Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A (2000) Enhancement of tumour necrosis factor alpha antitumour immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat Biotechnol 18: 1185–1190
Curnis F, Sacchi A, Corti A (2002) Improving chemotherapeutic drug penetration in tumours by vascular targeting and barrier alteration. J Clin Invest 110: 475–482
d’Arcy JA, Collins DJ, Padhani AR, Walker-Samuel S, Suckling J, Leach MO (2006) Informatics in Radiology (infoRAD): Magnetic Resonance Imaging Workbench: analysis and visualization of dynamic contrast-enhanced MR imaging data. Radiographics 26: 621–632
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8: 579–591
Gregorc V, Citterio C, Vitali G, Spreafico A, Scifo P, Borri A, Donadoni G, Rossoni G, Corti C, Caligaris-Cappio F, Del Maschio A, Esposito A, De Cobelli F, Dell’Acqua F, Troysi A, Bruzzi P, Lambiase A, Bordignon C (2010) Defining the optimal biological dose of NGR-hTNF, a selective vascular targeting agent, in advanced solid tumours. Eur J Cancer 46: 198–206
Fraker DL, Alexander HR, Pass HI (1995) Biologic therapy with TNF: systemic administration and isolation-perfusion. In Biologic Therapy of Cancer: Principles and Practice, De Vita V, Hellman S, Rosenberg S, (eds) pp 329–345. JB Lippincott Co: Philadelphia, Pennsylvania, USA
Jackson A, O’Connor JPB, Parker GJM, Jayson GC (2007) Imaging tumour vascular heterogeneity and angiogenesis using dynamic contrast-enhanced magnetic resonance Imaging. Clin Cancer Res 13: 3449–3459
Lejeune FJ, Lienard D, Matter M, Ruegg C (2006) Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun 6: 1–17
Lenz HJ (2007) Management and preparedness for infusion and hypersensitivity reactions. Oncologist 12: 601–609
O’Connor JPB, Jackson A, Geoff JM, Parker GJM, Roberts C, Jayson GC (2012) Dynamic contrast-enhanced MRI in clinical trials of antivascular therapies. Nat Rev Clin Oncol 9: 167–177
Rangel R, Sun Y, Guzman-Rojas L, Ozawa MG, Sun J, Giordano RJ (2007) Impaired angiogenesis in aminopeptidase N-null mice. Proc Natl Acad Sci USA 4588–4593
Sacchi A, Gasparri A, Gallo-Stampino C, Toma S, Curnis F, Corti A (2006) Synergistic antitumour activity of cisplatin, paclitaxel, and gemcitabine with tumour vasculature-targeted tumour necrosis factor-alpha. Clin Cancer Res 12: 175–182
Tofts PS, Brix G, Buckley DL, Evelhoch JL, Henderson E, Knopp MV, Henrik Larsson HBW, Lee TY, Mayr NA, Parker GJM, Port RE, Taylor J, Weisskoff RM (1999) Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: standardized quantities and symbols. J Magn Reson Imaging 10: 223–232
van Heeckeren WJ, Bhakta S, Ortis J (2006) Promise of new vascular-disrupting agents balanced with cardiac toxicity: is it time for oncologists to get to know their cardiologists? J Clin Oncol 24: 1485–1488
van Laarhoven HWM, Fiedler W, Desar IME, van Asten JJA, Sandrine Marreaud S, Lacombe D, Govaerts AS, Bogaerts J, Lasch P, Timmer-Bonte JNH, Lambiase A, Bordignon C, Punt CJA, Heerschap A, van Herpen CML (2010) Phase I clinical and magnetic resonance imaging study of the vascular agent NGR-hTNF in patients with advanced cancers (European Organization for Research and Treatment of Cancer Study 16041). Clin Cancer Res 16: 1315–1323
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zucali, P., Simonelli, M., De Vincenzo, F. et al. Phase I and pharmacodynamic study of high-dose NGR–hTNF in patients with refractory solid tumours. Br J Cancer 108, 58–63 (2013). https://doi.org/10.1038/bjc.2012.506
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/bjc.2012.506
Keywords
This article is cited by
-
ACO2 and ANPEP as novel prognostic markers for gallbladder squamous cell/adenosquamous carcinomas and adenocarcinomas
International Journal of Clinical Oncology (2020)
-
Anti-metastatic activity of the tumor vascular targeting agent NGR-TNF
Clinical & Experimental Metastasis (2015)
-
NGR-TNF, a novel vascular-targeting agent, does not induce cytokine recruitment of proangiogenic bone marrow-derived cells
British Journal of Cancer (2013)
-
Peptide-Mediated Targeting of Cytokines to Tumor Vasculature: The NGR-hTNF Example
BioDrugs (2013)
