
Abstract
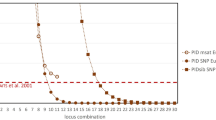
Genetic matching potentially provides a means to alleviate the effects of incomplete Mendelian randomization in population-based gene–disease association studies. We therefore evaluated the genetic-matched pair study design on the basis of genome-wide SNP data (309 790 markers; Affymetrix GeneChip Human Mapping 500K Array) from 2457 individuals, sampled at 23 different recruitment sites across Europe. Using pair-wise identity-by-state (IBS) as a matching criterion, we tried to derive a subset of markers that would allow identification of the best overall matching (BOM) partner for a given individual, based on the IBS status for the subset alone. However, our results suggest that, by following this approach, the prediction accuracy is only notably improved by the first 20 markers selected, and increases proportionally to the marker number thereafter. Furthermore, in a considerable proportion of cases (76.0%), the BOM of a given individual, based on the complete marker set, came from a different recruitment site than the individual itself. A second marker set, specifically selected for ancestry sensitivity using singular value decomposition, performed even more poorly and was no more capable of predicting the BOM than randomly chosen subsets. This leads us to conclude that, at least in Europe, the utility of the genetic-matched pair study design depends critically on the availability of comprehensive genotype information for both cases and controls.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
GAIN Collaborative Research Group Manolio TA, Rodriguez LL, Brooks L et al: New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet 32007; 9: 1045–1051.
Hirschhorn JN : Genetic approaches to studying common diseases and complex traits. Pediatr Res 2005; 57: 74R–77R.
The Wellcome Trust Case Control Consortium: Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678.
Luca D, Ringquist S, Klei L et al: On the use of general control samples for genome-wide association studies: genetic matching highlights causal variants. Am J Hum Genet 2008; 82: 453–463.
Davey Smith G, Ebrahim S : What can mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ 2005; 330: 1076–1079.
Devlin B, Roeder K : Genomic control for association studies. Biometrics 1999; 55: 997–1004.
Pritchard JK, Stephens M, Donnelly P : Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Wang WY, Barratt BJ, Clayton DG, Todd JA : Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 2005; 6: 109–118.
Wichmann HE, Gieger C, Illig T, MONICA/KORA_Study_Group: KORA-gen – resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 2005; 67: 26–30.
Lao O, Lu TT, Nothnagel M et al: Correlation between genetic and geographic structure in Europe. Curr Biol 2008; 18: 1241–1248.
R Development Core Team: R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, 2008.
Morton NE, Zhang W, Taillon-Miller P, Ennis S, Kwok PY, Collins A : The optimal measure of allelic association. Proc Natl Acad Sci USA 2001; 98: 5217–5221.
Wollstein A, Herrmann A, Wittig M et al: Efficacy assessment of SNP sets for genome-wide disease association studies. Nucleic Acids Res 2007; 35: e113.
Paschou P, Drineas P, Lewis J et al: Tracing sub-structure in the European American population with PCA-informative markers. PLoS Genet 2008; 4: e1000114+.
Paschou P, Ziv E, Burchard EG et al: PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet 2007; 3: 1672–1686.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Berry MW : Large scale singular value computations. Int J Supercomput Appl 1992; 6: 13–49.
Eaton JW : GNU Octave Manual. Network Theory Unlimited: Bristol, 2002.
Gansner ER, North SC : An open graph visualization system and its applications to software engineering. Softw Pract Exp 2000; 30: 1203–1233.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Kayser M, Liu F, Janssens AC et al: Three genome-wide association studies and a linkage analysis identify HERC2 as a human iris color gene. Am J Hum Genet 2008; 82: 411–423.
Duffy DL, Montgomery GW, Chen W et al: A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. Am J Hum Genet 2007; 80: 241–252.
Bersaglieri T, Sabeti PC, Patterson N et al: Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet 2004; 74: 1111–1120.
Voight BF, Kudaravalli S, Wen X, Pritchard JK : A map of recent positive selection in the human genome. PLoS Biol 2006; 4: e72.
Bauchet M, McEvoy B, Pearson LN et al: Measuring European population stratification with microarray genotype data. Am J Hum Genet 2007; 80: 948–956.
Price AL, Butler J, Patterson N et al: Discerning the ancestry of European Americans in genetic association studies. PLoS Genet 2008; 4: e236.
Seldin MF, Shigeta R, Villoslada P et al: European population substructure: clustering of northern and southern populations. PLoS Genet 2006; 2: e143.
Tian C, Hinds DA, Shigeta R et al: A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet 2007; 80: 1014–1023.
Heath SC, Gut IG, Brennan P et al: Investigation of the fine structure of European populations with applications to disease association studies. Eur J Hum Genet 2008; 16: 1413–1429.
Acknowledgements
All sample donors are gratefully acknowledged for their participation. We thank the following colleagues for their help and support: J Kooner and J Chambers of the LOLIPOP study and D Waterworth, V Mooser, G Waeber and P Vollenweider of the CoLaus study for providing access to their collections through the GlaxoSmithKline-sponsored Population Reference Sample (POPRES) project; K King for preparing the POPRES data; M Simoons, E Sijbrands, A van Belkum, J Laven, J Lindemans, E Knipers and B Stricker for their financial contribution to the Rotterdam study; P Arp, M Jhamai, W van IJken and R van Schaik for generating the Rotterdam study dataset; T Meitinger, P Lichtner, G Eckstein and all genotyping staff at the Helmholtz Zentrum München for generating the KORA study dataset; H von Eller-Eberstein for providing access to the PopGen data; R Borup, C Schjerling, H Ullum, E Haastrup and numerous colleagues at the Copenhagen University Hospital Blood Bank for making the Danish data available; and S Brauer for DNA sample management. We also wish to thank Affymetrix for making the GeneChip Human Mapping 500K Array genotypes of the CEPH-CEU trios publicly available, and the Centre d’Etude du Polymorphisme Humain (CEPH) for the original sample collection. This work was supported by the Netherlands Forensic Institute (M Ka), Affymetrix (M Ka and M Kr), the German National Genome Research Network and the German Federal Ministry of Education and Research (H-EW, SS, M Kr and PN); the Helmholtz Zentrum München – German Research Center for Environmental Health, Neuherberg and the Munich Center of Health Sciences as part of LMUinnovativ (H-EW), the Netherlands Organization for Scientific Research (AGU: NWO 175.010.2005.011), the European Commission (AGU: GEFOS; 201865, AS: LD Europe; QLG2-CT-2001-00916); the Czech Ministry of Health (MM: VZFNM 00064203 and IGA NS/9488-3), Helse-Vest, Regional Health Authority Norway (LAB), the Swedish National Board of Forensic Medicine (GH: RMVFoU 99:22, 02:20) and the Academy of Finland (AS: 80578, OMLL, JP: 109265 and 111713). None of the funding organization had any influence on the design, conduct or conclusions of the study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Lu, T., Lao, O., Nothnagel, M. et al. An evaluation of the genetic-matched pair study design using genome-wide SNP data from the European population. Eur J Hum Genet 17, 967–975 (2009). https://doi.org/10.1038/ejhg.2008.266
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2008.266
Keywords
This article is cited by
-
The more the merrier? How a few SNPs predict pigmentation phenotypes in the Northern German population
European Journal of Human Genetics (2016)
