
Abstract
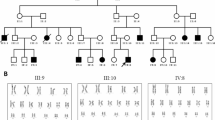
Split hand-split foot malformation or ectrodactyly is a heterogeneous congenital defect of digit formation. The aim of this study is the mapping of the breakpoints and a detailed molecular characterization of the candidate genes for an isolated and syndromic form of ectrodactyly, both associated with de novo apparently balanced chromosome translocations involving the same chromosome 2 band, [t(2;11)(q14.2;q14.2)] and [t(2;4)(q14.1;q35)], respectively. Breakpoints were mapped by fluorescence in situ hybridization using bacterial artificial chromosome clones. Where possible, these breakpoints were further delimited. Candidate genes were screened for pathogenic mutations and the expression levels of two of them analysed. The isolated bilateral split foot malformation-associated chromosome 2 breakpoint was localized at 120.9 Mb, between the two main candidate genes, encoding GLI-Kruppel family member GLI2 and inhibin-βB. The second breakpoint associated with holoprosencephaly, hypertelorism and ectrodactyly syndrome was mapped 2.5 Mb proximal at 118.4 Mb and the candidate genes identified from this region were the insulin-induced protein 2 and the homeobox protein engrailed-1. No clear pathogenic mutations were identified in any of these genes. The breakpoint between INHBB and GLI2 coincides with a previously identified translocation breakpoint associated with ectrodactyly. We propose a mechanism by which translocations in the 2q14.1–q14.2 region disrupt the specific arrangement of long-range regulatory elements that control the tight quantitative spatiotemporal expression of one or more genes from the breakpoint region.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Duijf PHG, van Bokhoven H, Brunner HG : Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet 2003; 12: R51–R60.
Van Bokhoven H, Brunner HG : Splitting p63. Am J Hum Genet 2002; 71: 1–13.
Kjaer KW, Hansen L, Schwabe GC et al: Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J Med Genet 2005; 42: 292–298.
Bernardini L, Palka C, Ceccarini C et al: Complex rearrangement of chromosome 7q21.13-q22.1 confirms the ectrodactyly-deafness locus and suggests new candidate genes. Am J Med Genet 2008; 146A: 238–244.
de Mollerat XJ, Gurrieri F, Morgan CT et al: A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 2003; 12: 1959–1971.
Goodman FR, Majewski F, Collins AL, Scambler PJ : A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly. Am J Hum Genet 2002; 70: 547–555.
Gurnett CA, Dobbs MB, Nordsieck EJ et al: Evidence for an additional locus for split hand/foot malformation in chromosome region 8q21.11-q22.3. Am J Med Genet 2006; 140A: 1744–1748.
Corona-Rivera A, Corona-Rivera JR, Bobadilla-Morales L, García-Cobian TA, Corona-Rivera E : Holoprosencephaly, hypertelorism, and ectrodactyly in a boy with an apparently balanced de novo t(2;4) (q14.2;q35). Am J Med Genet 2000; 90: 423–426.
König R, Beeg T, Tariverdian G, Scheffer H, Bitter K : Holoprosencephaly, bilateral cleft lip and palate and ectrodactyly: another case and follow up. Clin Dysmorphol 2003; 12: 221–225.
Naveed M, Nath SK, Gaines M et al: Genomewide linkage scan for split-hand/foot malformation with long-bone deficiency in a large Arab family identifies two novel susceptibility loci on chromosomes 1q42.2-q43 and 6q14.1. Am J Hum Genet 2007; 80: 105–111.
Babbs C, Heller R, Everman DB et al: A new locus for split hand/foot malformation with long bone deficiency (SHFLD) at 2q14.2 identified from a chromosome translocation. Hum Genet 2007; 122: 191–199.
Van Roy N, Jauch A, Van Gele M et al: Comparative genomic hybridization analysis of human neuroblastomas: detection of distal lp deletions and further molecular genetic characterization of neuroblastoma cell lines. Cancer Genet Cytogenet 1997; 97: 135–142.
David D, Cardoso J, Marques B et al: Molecular characterization of a familial translocation implicates disruption of HDAC9 and possible position effect on TGFbeta2 in the pathogenesis of Peters’ anomaly. Genomics 2003; 81: 489–503.
David D, Ribeiro S, Ferrão L, Gago T, Campos M, Crespo F : Molecular basis of inherited antithrombin deficiency in Portuguese families: identification of underlying molecular defects and screening for additional thrombotic risk factors. Am J Hematol 2004; 76: 163–171.
de Brouwer AP, van Bokhoven H, Kremer H : Comparison of 12 reference genes for normalization of gene expression levels in Epstein-Barr virus-transformed lymphoblastoid cell lines and fibroblasts. Mol Diagn Ther 2006; 10: 197–204.
Roessler E, Du YZ, Mullor JL et al: Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA 2003; 100: 13424–13429.
Kleinjan DA, van Heyningen V : Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 2005; 76: 8–32.
Koebernick K, Pieler T : Gli-type zinc finger proteins as bipotential transducers of Hedgehog signaling. Differentiation 2002; 70: 69–76.
Miao D, Liu H, Plut P et al: Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp Cell Res 2004; 294: 210–222.
Zhao M, Qiao M, Harris SE, Chen D, Oyajobi BO, Mundy GR : The zinc finger transcription factor Gli2 mediates bone morphogenetic protein 2 expression in osteoblasts in response to hedgehog signaling. Mol Cell Biol 2006; 26: 6197–6208.
Nieuwenhuis E, Hui C-C : Hedgehog signaling and congenital malformations. Clin Genet 2004; 67: 193–208.
Thompson TB, Cook RW, Chapman SC, Jardetzky TS, Woodruff TK : Beta A versus beta B: is it merely a matter of expression? Mol Cell Endocrinol 2004; 225: 9–17.
Merino R, Macias D, Gañan Y et al: Control of digit formation by activin signalling. Development 1999; 126: 2161–2170.
Schrewe H, Gendron-Maguire M, Harbison ML, Gridley T : Mice homozygous for a null mutation of activin beta B are viable and fertile. Mech Dev 1994; 47: 43–51.
Cervino AC, Li G, Edwards S et al: Integrating QTL and high-density SNP analyses in mice to identify Insig2 as a susceptibility gene for plasma cholesterol levels. Genomics 2005; 86: 505–517.
Yabe D, Brown MS, Goldstein JL : Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA 2002; 99: 12753–12758.
Cooper MK, Wassif CA, Krakowiak PA et al: A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet 2003; 33: 508–513.
Wurst W, Auerbach AB, Joyner AL : Multiple developmental defects in Engrailed-1 mutant mice: an early mid-hindbrain deletion and patterning defects in forelimbs and sternum. Development 1994; 120: 2065–2075.
Loomis CA, Kimmel RA, Tong C-X, Michaud J, Joyner AL : Analysis of the genetic pathway leading to formation of ectopic apical ectodermal ridges in mouse Engrailed-1 mutant limbs. Development 1998; 125: 1137–1148.
Barber JC, Maloney VK, Bewes B, Wakeling E : Deletions of 2q14 that include the homeobox engrailed 1 (EN1) transcription factor are compatible with a normal phenotype. Eur J Hum Genet 2006; 14: 739–743.
Lettice LA, Heaney SJH, Purdie LA et al: A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 2003; 12: 1725–1735.
Acknowledgements
We thank Purificação Tavares for pre-natal diagnosis of the translocation; Anabela L Dias for maintaining the LCL; Catarina Reis for contributing to the screening of the 5′UTR of INHBB; Sara Malveiro for contributing to fine mapping of the chromosome 2 breakpoint; Emine Bolat for quantitative RT-PCR analysis; Paula Borralho and Ricardo Laurini, for morphopathological evaluation of the abortus; and Raoul Hennekam for contributing patient material. This project was partially supported by Fundação para a Ciência e a Tecnologia research Grant POCTI/MGI/38649/2001, by Programa de Financiamento Plurianual do CIGMH and by Fundo Comunitário Europeu FEDER, and the European Union Sixth Framework program EpiStem project (LSHB-CT-2005-019067).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
David, D., Marques, B., Ferreira, C. et al. Characterization of two ectrodactyly-associated translocation breakpoints separated by 2.5 Mb on chromosome 2q14.1–q14.2. Eur J Hum Genet 17, 1024–1033 (2009). https://doi.org/10.1038/ejhg.2009.2
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2009.2
Keywords
This article is cited by
-
Molecular characterization of a rare analphoid supernumerary marker chromosome derived from 7q35 → qter: a case report
Molecular Cytogenetics (2016)
-
Co-segregation of trichorhinophalangeal syndrome with a t(8;13)(q23.3;q21.31) familial translocation that appears to increase TRPS1 gene expression
Human Genetics (2013)
