
Abstract
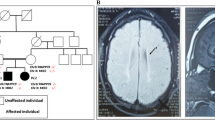
Heterozygous in-frame mutations (p.E2207del and p.R2308_M2309dup) in the α-II subunit of spectrin (SPTAN1) were recently identified in two patients with intellectual disability (ID), infantile spasms (IS), hypomyelination, and brain atrophy. These mutations affected the C-terminal domain of the protein, which contains the nucleation site of the α/β spectrin heterodimer. By screening SPTAN1 in 95 patients with idiopathic ID, we found a de novo in-frame mutation (p.Q2202del) in the same C-terminal domain in a patient with mild generalized epilepsy and pontocerebellar atrophy, but without IS, hypomyelination, or other brain structural defects, allowing us to define the core phenotype associated with these C-terminal SPTAN1 mutations. We also found a de novo missense variant (p.R566P) of unclear clinical significance in a patient with non-syndromic ID. These two mutations induced different patterns of aggregation between spectrin subunits in transfected neuronal cell lines, providing a paradigm for the classification of candidate variants.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Bennett V, Healy J : Organizing the fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol Med 2008; 14: 28–36.
Baines AJ : Evolution of spectrin function in cytoskeletal and membrane networks. Biochem Soc Trans 2009; 37: 796–803.
Ikeda Y, Dick KA, Weatherspoon MR et al: Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 2006; 38: 184–190.
Saitsu H, Tohyama J, Kumada T et al: Dominant-negative mutations in alpha-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet 2010; 86: 881–891.
Speicher DW, Weglarz L, DeSilva TM : Properties of human red cell spectrin heterodimer (side-to-side) assembly and identification of an essential nucleation site. J Biol Chem 1992; 267: 14775–14782.
Conrad DF, Keebler JE, DePristo MA et al: Variation in genome-wide mutation rates within and between human families. Nat Genet 2011; 43: 712–714.
Hamdan FF, Gauthier J, Araki Y et al: Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 2011; 88: 306–316.
Vissers LE, de Ligt J, Gilissen C et al: A de novo paradigm for mental retardation. Nat Genet 2010; 42: 1109–11012.
Saitsu H, Kato M, Mizuguchi T et al: De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008; 40: 782–788.
Ng PC, Henikoff S : SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814.
Sunyaev S, Ramensky V, Koch I, Lathe 3rd W, Kondrashov AS, Bork P : Prediction of deleterious human alleles. Hum Mol Genet 2001; 10: 591–597.
Acknowledgements
This study is supported by grants from the Canadian Institute of Health Research (CIHR) (J Michaud, G Rouleau, J-C Lacaille), Réseau de Génétique Médicale Appliquée (RMGA)/Fonds de la Recherche en Santé du Québec (FRSQ) (J Michaud), Genome Canada and Genome Quebec and co-funding by Université de Montréal for the Synapse to diseases (S2D) project (G Rouleau), the Ministry of Health, Labour and Welfare (H Saitsu, N Matsumoto), the Japan Science and Technology Agency (N Matsumoto), a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (N Matsumoto), and a Grant-in-Aid for Young Scientist from the Japan Society for the Promotion of Science (H Saitsu). J Michaud is the recipient of a Clinical Investigatorship Award of the CIHR (Institute of Genetics) and of a Senior Scientist Award from FRSQ. G Rouleau holds the Canada Research Chair and a Jeanne-et-J-Louis-Levesque Chair for the Genetics of Brain Diseases. We are grateful for the dedicated work of members of the S2D team (CHUM Notre-Dame Hospital Research Center, Montreal), including management (Claude Marineau and Ronald Lafrenière) and bioinformatics (Edouard Henrion and Ousmane Diallo). We are thankful for the efforts of the members of McGill University and Génome Québec Innovation Centre Sequencing and Bioinformatics groups.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Hamdan, F., Saitsu, H., Nishiyama, K. et al. Identification of a novel in-frame de novo mutation in SPTAN1 in intellectual disability and pontocerebellar atrophy. Eur J Hum Genet 20, 796–800 (2012). https://doi.org/10.1038/ejhg.2011.271
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2011.271
Keywords
This article is cited by
-
SPTAN1 variants likely cause autosomal recessive complicated hereditary spastic paraplegia
Journal of Human Genetics (2022)
-
SPTAN1 encephalopathy: distinct phenotypes and genotypes
Journal of Human Genetics (2015)
