
Abstract
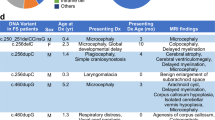
The Forkhead box G1 (FOXG1) gene has been implicated in severe Rett-like phenotypes. It encodes the Forkhead box protein G1, a winged-helix transcriptional repressor critical for forebrain development. Recently, the core FOXG1 syndrome was defined as postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and dysgenesis of the corpus callosum. We present seven additional patients with a severe Rett-like neurodevelopment disorder associated with de novo FOXG1 point mutations (two cases) or 14q12 deletions (five cases). We expand the mutational spectrum in patients with FOXG1-related encephalopathies and precise the core FOXG1 syndrome phenotype. Dysgenesis of the corpus callosum and dyskinesia are not always present in FOXG1-mutated patients. We believe that the FOXG1 gene should be considered in severely mentally retarded patients (no speech-language) with severe acquired microcephaly (−4 to−6 SD) and few clinical features suggestive of Rett syndrome. Interestingly enough, three 14q12 deletions that do not include the FOXG1 gene are associated with phenotypes very reminiscent to that of FOXG1-mutation-positive patients. We physically mapped a putative long-range FOXG1-regulatory element in a 0.43 Mb DNA segment encompassing the PRKD1 locus. In fibroblast cells, a cis-acting regulatory sequence located more than 0.6 Mb away from FOXG1 acts as a silencer at the transcriptional level. These data are important for clinicians and for molecular biologists involved in the management of patients with severe encephalopathies compatible with a FOXG1-related phenotype.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Le Guen T, Fichou Y, Nectoux J et al. A missense mutation within the fork-head domain of the forkhead box G1 gene (FOXG1) affects its nuclear localization. Hum Mutat 2011; 32: E2026–E2035.
Philippe C, Amsallem D, Francannet C et al. Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. J Med Genet 2010; 47: 59–65.
Mencarelli MA, Spanhol-Rosseto A, Artuso R et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet 2010; 47: 49–53.
Ariani F, Hayek G, Rondinella D et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008; 83: 89–93.
Van der Aa N, Van den Bergh M, Ponomarenko N, Verstraete L, Ceulemans B, Storm K : Analysis of FOXG1 is highly recommended in male and female patients with rett syndrome. Mol Syndromol 2011; 1: 290–293.
Kortum F, Das S, Flindt M et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet 2011; 47: 49–53.
Le Guen T, Bahi-Buisson N, Nectoux J et al. A FOXG1 mutation in a boy with congenital variant of Rett syndrome. Neurogenetics 2011; 12: 1–8.
Mencarelli MA, Kleefstra T, Katzaki E et al. 14q12 Microdeletion syndrome and congenital variant of Rett syndrome. Eur J Med Genet 2009; 52: 148–152.
Jacob FD, Ramaswamy V, Andersen J, Bolduc FV : Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet 2009; 17: 1577–1581.
Striano P, Paravidino R, Sicca F et al. West syndrome associated with 14q12 duplications harboring FOXG1. Neurology 2011; 76: 1600–1602.
Brunetti-Pierri N, Paciorkowski AR, Ciccone R et al. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet 2011; 19: 102–107.
Yeung A, Bruno D, Scheffer IE et al. 4.45 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur J Med Genet 2009; 52: 440–442.
Tohyama J, Yamamoto T, Hosoki K et al. West syndrome associated with mosaic duplication of FOXG1 in a patient with maternal uniparental disomy of chromosome 14. Am J Med Genet A 2011; 155A: 2584–2588.
Shoichet SA, Kunde SA, Viertel P et al. Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet 2005; 117: 536–544.
Quenard A, Yilmaz S, Fontaine H et al. Deleterious mutations in exon 1 of MECP2 in Rett syndrome. Eur J Med Genet 2006; 49: 313–322.
Bonnet C, Leheup B, Beri M, Philippe C, Gregoire MJ, Jonveaux P : Aberrant GRIA3 transcripts with multi-exon duplications in a family with X-linked mental retardation. Am J Med Genet A 2009; 149A: 1280–1289.
Bahi-Buisson N, Nectoux J, Girard B et al. Revisiting the phenotype associated with FOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics 2010; 11: 241–249.
Takahashi S, Matsumoto N, Okayama A et al. FOXG1 mutations in Japanese patients with the congenital variant of Rett syndrome. Clin Genet 2011; e-pub ahead of print 1 December; doi:10.1111/j.1399-0004.2011.01819.x.
Bisgaard AM, Kirchhoff M, Tumer Z et al. Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am J Med Genet A 2006; 140: 2180–2187.
Papa FT, Mencarelli MA, Caselli R et al. A 3 Mb deletion in 14q12 causes severe mental retardation, mild facial dysmorphisms and Rett-like features. Am J Med Genet A 2008; 146A: 1994–1998.
Roche-Martinez A, Gerotina E, Armstrong-Moron J, Sans-Capdevila O, Pineda M : [FOXG1, a new gene responsible for the congenital form of Rett syndrome.]. Rev Neurol 2011; 52: 597–602.
De Filippis R, Pancrazi L, Bjorgo K et al. Expanding the phenotype associated with FOXG1 mutations and in vivo FoxG1 chromatin-binding dynamics. Clin Genet 2011; e-pub ahead of print 17 November. doi:10.1111/j.1399-0004.2011.01810.x.
Amor DJ, Burgess T, Tan TY, Pertile MD : Questionable pathogenicity of FOXG1 duplication. Eur J Hum Genet 2012; 20: 595–596.
Brunetti-Pierri N, Cheung SW, Stankiewicz P : Reply to Amor et al. Eur J Hum Genet 2012; 20: 597.
Huynh QK, McKinsey TA : Protein kinase D directly phosphorylates histone deacetylase 5 via a random sequential kinetic mechanism. Arch Biochem Biophys 2006; 450: 141–148.
Kisfalvi K, Hurd C, Guha S, Rozengurt E : Induced overexpression of protein kinase D1 stimulates mitogenic signaling in human pancreatic carcinoma PANC-1 cells. J Cell Physiol 2010; 223: 309–316.
Czondor K, Ellwanger K, Fuchs YF et al. Protein kinase D controls the integrity of Golgi apparatus and the maintenance of dendritic arborization in hippocampal neurons. Mol Biol Cell 2009; 20: 2108–2120.
Calalb MB, McKinsey TA, Newkirk S, Huynh K, Sucharov CC, Bristow MR : Increased phosphorylation-dependent nuclear export of class II histone deacetylases in failing human heart. Clin Transl Sci 2009; 2: 325–332.
Benko S, Fantes JA, Amiel J et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet 2009; 41: 359–364.
Williamson SL, Christodoulou J : Rett syndrome: new clinical and molecular insights. Eur J Hum Genet 2006; 14: 896–903.
Acknowledgements
We gratefully acknowledge clinicians who have referred patients for MECP2/CDKL5/FOXG1 analyses, and more particularly Professor L Olivier-Faivre, Dr F Giuliano, Dr S Julia, Dr V Layet, and Dr A Roubertie. We thank MD Devignes (LORIA, Vandoeuvre les Nancy, France) for help with bioinformatic analyses. This work was supported by grants (CPRC 04.9582, 2009) from the Centre Hospitalier Universitaire de Nancy and from the Association Française du Syndrome de Rett (AFSR). LA is funded by a grant MESR (Ministère de l'Enseignement Supérieur et de la Recherche).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Allou, L., Lambert, L., Amsallem, D. et al. 14q12 and severe Rett-like phenotypes: new clinical insights and physical mapping of FOXG1-regulatory elements. Eur J Hum Genet 20, 1216–1223 (2012). https://doi.org/10.1038/ejhg.2012.127
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2012.127
Keywords
This article is cited by
-
Expanding genotype–phenotype correlations in FOXG1 syndrome: results from a patient registry
Orphanet Journal of Rare Diseases (2023)
-
Learning representations of chromatin contacts using a recurrent neural network identifies genomic drivers of conformation
Nature Communications (2022)
-
Prenatal diagnosis of maternal partial trisomy 9p23p24.3 and 14q11.2q21.3 in a fetus: a case report
Molecular Cytogenetics (2020)
-
A novel 14q13.1–21.1 deletion identified by CNV-Seq in a patient with brain-lung-thyroid syndrome, tooth agenesis and immunodeficiency
Molecular Cytogenetics (2019)
-
Foxg1 deletion impairs the development of the epithalamus
Molecular Brain (2018)
