
Abstract
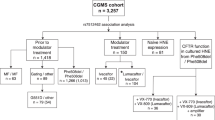
There is growing evidence that the great phenotypic variability in patients with cystic fibrosis (CF) not only depends on the genotype, but apart from a combination of environmental and stochastic factors predominantly also on modifier gene effects. It has been proposed that genes interacting with CF transmembrane conductance regulator (CFTR) and epithelial sodium channel (ENaC) are potential modifiers. Therefore, we assessed the impact of single-nucleotide polymorphisms (SNPs) of several of these interacters on CF disease outcome. SNPs that potentially alter gene function were genotyped in 95 well-characterized p.Phe508del homozygous CF patients. Linear mixed-effect model analysis was used to assess the relationship between sequence variants and the repeated measurements of lung function parameters. In total, we genotyped 72 SNPs in 10 genes. Twenty-five SNPs were used for statistical analysis, where we found strong associations for one SNP in PPP2R4 with the lung clearance index (P≤0.01), the specific effective airway resistance (P≤0.005) and the forced expiratory volume in 1 s (P≤0.005). In addition, we identified one SNP in SNAP23 to be significantly associated with three lung function parameters as well as one SNP in PPP2R1A and three in KRT19 to show a significant influence on one lung function parameter each. Our findings indicate that direct interacters with CFTR, such as SNAP23, PPP2R4 and PPP2R1A, may modify the residual function of p.Phe508del-CFTR while variants in KRT19 may modulate the amount of p.Phe508del-CFTR at the apical membrane and consequently modify CF disease.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Riordan JR, Rommens JM, Kerem B et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–1073.
Kerem B, Rommens JM, Buchanan JA et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073–1080.
Collins FS : Cystic fibrosis: molecular biology and therapeutic implications. Science 1992; 256: 774–779.
Rosenstein BJ, Cutting GR : The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr 1998; 132: 589–595.
Knowles MR, Durie PR : What is cystic fibrosis? N Engl J Med 2002; 347: 439–442.
Corey M, Edwards L, Levison H, Knowles M : Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr 1997; 131: 809–814.
Gustafsson PM, Aurora P, Lindblad A : Evaluation of ventilation maldistribution as an early indicator of lung disease in children with cystic fibrosis. Eur Respir J 2003; 22: 972–979.
Kraemer R, Blum A, Schibler A, Ammann RA, Gallati S : Ventilation inhomogeneities in relation to standard lung function in patients with cystic fibrosis. Am J Respir Crit Care Med 2005; 171: 371–378.
Kraemer R, Baldwin DN, Ammann RA, Frey U, Gallati S : Progression of pulmonary hyperinflation and trapped gas associated with genetic and environmental factors in children with cystic fibrosis. Respir Res 2006; 7: 138.
Kraemer R, Latzin P, Pramana I, Ballinari P, Gallati S, Frey U : Long-term gas exchange characteristics as markers of deterioration in patients with cystic fibrosis. Respir Res 2009; 10: 106.
Aurora P, Bush A, Gustafsson P et al. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am J Respir Crit Care Med 2005; 171: 249–256.
Kraemer R, Birrer P, Liechti-Gallati S : Genotype-phenotype association in infants with cystic fibrosis at the time of diagnosis. Pediatr Res 1998; 44: 920–926.
Ranganathan SC, Stocks J, Dezateux C et al. The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med 2004; 169: 928–933.
Konstan MW, Hilliard KA, Norvell TM, Berger M : Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med 1994; 150: 448–454.
Ranganathan SC, Dezateux C, Bush A et al. Airway function in infants newly diagnosed with cystic fibrosis. Lancet 2001; 358: 1964–1965.
Vanscoy LL, Blackman SM, Collaco JM et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med 2007; 175: 1036–1043.
Buranawuti K, Boyle MP, Cheng S et al. Variants in mannose-binding lectin and tumour necrosis factor alpha affect survival in cystic fibrosis. J Med Genet 2007; 44: 209–214.
Bremer LA, Blackman SM, Vanscoy LL et al. Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum Mol Genet 2008; 17: 2228–2237.
Gu Y, Harley IT, Henderson LB et al. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature 2009; 458: 1039–1042.
Hillian AD, Londono D, Dunn JM et al. Modulation of cystic fibrosis lung disease by variants in interleukin-8. Genes Immun 2008; 9: 501–508.
Darrah R, McKone E, O’Connor C et al. EDNRA variants associate with smooth muscle mRNA levels, cell proliferation rates, and cystic fibrosis pulmonary disease severity. Physiol Genomics 2010; 41: 71–77.
Wright FA, Strug LJ, Doshi VK et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 2011; 43: 539–546.
Cormet-Boyaka E, Di A, Chang SY et al. CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex. Proc Natl Acad Sci USA 2002; 99: 12477–12482.
Tang BL, Gee HY, Lee MG : The cystic fibrosis transmembrane conductance regulator's expanding SNARE interactome. Traffic 2011; 12: 364–371.
Le Drevo MA, Benz N, Kerbiriou M et al. Annexin A5 increases the cell surface expression and the chloride channel function of the DeltaF508-cystic fibrosis transmembrane regulator. Biochim Biophys Acta 2008; 1782: 605–614.
Faria D, Dahimene S, Alessio L et al. Effect of Annexin A5 on CFTR: regulated traffic or scaffolding? Mol Membr Biol 2011; 28: 14–29.
Luo J, Pato MD, Riordan JR, Hanrahan JW : Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am J Physiol 1998; 274: C1397–C1410.
Thelin WR, Kesimer M, Tarran R et al. The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J Biol Chem 2005; 280: 41512–41520.
Vastiau A, Cao L, Jaspers M et al. Interaction of the protein phosphatase 2A with the regulatory domain of the cystic fibrosis transmembrane conductance regulator channel. FEBS Lett 2005; 579: 3392–3396.
Rotin D, Staub O : Role of the ubiquitin system in regulating ion transport. Pflugers Arch 2011; 461: 1–21.
Caohuy H, Jozwik C, Pollard HB : Rescue of DeltaF508-CFTR by the SGK1/Nedd4-2 signaling pathway. J Biol Chem 2009; 284: 25241–25253.
Myerburg MM, McKenna EE, Luke CJ, Frizzell RA, Kleyman TR, Pilewski JM : Prostasin expression is regulated by airway surface liquid volume and is increased in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 2008; 294: L932–L941.
Koulov AV, Lapointe P, Lu B et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol Biol Cell 2010; 21: 871–884.
Harada K, Okiyoneda T, Hashimoto Y et al. Calreticulin negatively regulates the cell surface expression of cystic fibrosis transmembrane conductance regulator. J Biol Chem 2006; 281: 12841–12848.
Salas PJ, Rodriguez ML, Viciana AL, Vega-Salas DE, Hauri HP : The apical submembrane cytoskeleton participates in the organization of the apical pole in epithelial cells. J Cell Biol 1997; 137: 359–375.
Sun F : Poster Session Abstracts. Pediatric Pulmology 2009; 44: 213–435.
Kraemer R, Meister B : Fast real-time moment-ratio analysis of multibreath nitrogen washout in children. J Appl Physiol 1985; 59: 1137–1144.
Kraemer R, Zehnder M, Meister B : Intrapulmonary gas distribution in healthy children. Respir Physiol 1986; 65: 127–137.
Zapletal A, Samanek M, Paul T : Lung function in Children and Adolescents: Methods, Reference Values. Basel: Karger, 1987.
Liechti-Gallati S, Schneider V, Neeser D, Kraemer R : Two buffer PAGE system-based SSCP/HD analysis: a general protocol for rapid and sensitive mutation screening in cystic fibrosis and any other human genetic disease. Eur J Hum Genet 1999; 7: 590–598.
Excoffier L, Lischer HE : Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10: 564–567.
Adzhubei IA, Schmidt S, Peshkin L et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Ng PC, Henikoff S : Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C : Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
Piva F, Giulietti M, Burini AB, Principato G : SpliceAid 2: a database of human splicing factors expression data and RNA target motifs. Hum Mutat 2012; 33: 81–85.
Sharma M, Benharouga M, Hu W, Lukacs GL : Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem 2001; 276: 8942–8950.
de Moor CH, Meijer H, Lissenden S : Mechanisms of translational control by the 3' UTR in development and differentiation. Semin Cell Dev Biol 2005; 16: 49–58.
Skoko N, Baralle M, Buratti E, Baralle FE : The pathological splicing mutation c.6792C>G in NF1 exon 37 causes a change of tenancy between antagonistic splicing factors. FEBS Lett 2008; 582: 2231–2236.
Acknowledgements
We are indebted to the patients and their families as well as to Professor Martin H Schöni, Professor Dr N Regamey, Dr Carmen Casaulta and the entire nursing staff of the Bernese Cystic Fibrosis Clinic for their contribution in collecting clinical data and the samples for genetic analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Gisler, F., von Kanel, T., Kraemer, R. et al. Identification of SNPs in the cystic fibrosis interactome influencing pulmonary progression in cystic fibrosis. Eur J Hum Genet 21, 397–403 (2013). https://doi.org/10.1038/ejhg.2012.181
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2012.181
Keywords
This article is cited by
-
Discrimination of single-point mutations in unamplified genomic DNA via Cas9 immobilized on a graphene field-effect transistor
Nature Biomedical Engineering (2021)
-
Clinical and molecular characterization of the potential CF disease modifier syntaxin 1A
European Journal of Human Genetics (2013)
-
Generation of Lung Epithelium from Pluripotent Stem Cells
Current Pathobiology Reports (2013)
