
Abstract
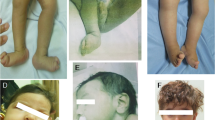
A chromosomal balanced translocation disrupting the MED13L (Mediator complex subunit13-like) gene, encoding a subunit of the Mediator complex, was previously associated with transposition of the great arteries (TGA) and intellectual disability (ID), and led to the identification of missense mutations in three patients with isolated TGA. Recently, a homozygous missense mutation in MED13L was found in two siblings with non-syndromic ID from a consanguineous family. Here, we describe for the first time, three patients with copy number changes affecting MED13L and delineate a recognizable MED13L haploinsufficiency syndrome. Using high resolution molecular karyotyping, we identified two intragenic de novo frameshift deletions, likely resulting in haploinsufficiency, in two patients with a similar phenotype of hypotonia, moderate ID, conotruncal heart defect and facial anomalies. In both, Sanger sequencing of MED13L did not reveal any pathogenic mutation and exome sequencing in one patient showed no evidence for a non-allelic second hit. A further patient with hypotonia, learning difficulties and perimembranous VSD showed a 1 Mb de novo triplication in 12q24.2, including MED13L and MAP1LC3B2. Our findings show that MED13L haploinsufficiency in contrast to the previously observed missense mutations cause a distinct syndromic phenotype. Additionally, a MED13L copy number gain results in a milder phenotype. The clinical features suggesting a neurocristopathy may be explained by animal model studies indicating involvement of the Mediator complex subunit 13 in neural crest induction.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Sato S, Tomomori-Sato C, Parmely TJ et al: A set of consensus mammalian mediator subunits identified by multidimensional protein identification technology. Mol Cell 2004; 14: 685–691.
Muncke N, Jung C, Rudiger H et al: Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (transposition of the great arteries). Circulation 2003; 108: 2843–2850.
Musante L, Bartsch O, Ropers HH, Kalscheuer VM : cDNA cloning and characterization of the human THRAP2 gene which maps to chromosome 12q24, and its mouse ortholog Thrap2. Gene 2004; 332: 119–127.
Risheg H, Graham JM, Clark RD et al: A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet 2007; 39: 451–453.
Schwartz CE, Tarpey PS, Lubs HA et al: The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet 2007; 44: 472–477.
Boutry-Kryza N, Labalme A, Till M et al: An 800 kb deletion at 17q23.2 including the MED13 (THRAP1) gene, revealed by aCGH in a patient with a SMC 17p. Am J Med Genet A 2012; 158A: 400–405.
Kaufmann R, Straussberg R, Mandel H et al: Infantile cerebral and cerebellar atrophy is associated with a mutation in the MED17 subunit of the transcription preinitiation Mediator complex. Am J Hum Genet 2010; 87: 667–670.
Hashimoto S, Boissel S, Zarhrate M et al: MED23 mutation links intellectual disability to dysregulation of immediate early gene expression. Science 2011; 333: 1161–1163.
Leal A, Huehne K, Bauer F et al: Identification of the variant Ala335Val of MED25 as responsible for CMT2B2: molecular data, functional studies of the SH3 recognition motif and correlation between wild-type MED25 and PMP22 RNA levels in CMT1A animal models. Neurogenetics 2009; 10: 275–287.
Najmabadi H, Hu H, Garshasbi M et al: Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478: 57–63.
Yobb TM, Somerville MJ, Willatt L et al: Microduplication and triplication of 22q11.2: a highly variable syndrome. Am J Hum Genet 2005; 76: 865–876.
Ruiter M, Koolen DA, Pfundt R et al: A novel 2.3 Mb microduplication of 12q24.21q24.23 detected by genome-wide tiling-path resolution array comparative genomic hybridization in a girl with syndromic mental retardation. Clin Dysmorphol 2006; 15: 133–137.
Wong KK, deLeeuw RJ, Dosanjh NS et al: A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet 2007; 80: 91–104.
Wang JC, Walker A, Blackwell TK, Yamamoto KR : The Caenorhabditis elegans ortholog of TRAP240, CeTRAP240/let-19, selectively modulates gene expression and is essential for embryogenesis. J Biol Chem 2004; 279: 29270–29277.
Lim J, Lee OK, Hsu YC, Singh A, Choi KW : Drosophila TRAP230/240 are essential coactivators for Atonal in retinal neurogenesis. Dev Biol 2007; 308: 322–330.
Ito M, Okano HJ, Darnell RB, Roeder RG : The TRAP100 component of the TRAP/Mediator complex is essential in broad transcriptional events and development. EMBO J 2002; 21: 3464–3475.
Conaway RC, Conaway JW : Function and regulation of the Mediator complex. Curr Opin Genet Dev 2011; 21: 225–230.
Yoda A, Kouike H, Okano H, Sawa H : Components of the transcriptional Mediator complex are required for asymmetric cell division in C. elegans. Development 2005; 132: 1885–1893.
Janody F, Treisman JE : Requirements for Mediator complex subunits distinguish three classes of notch target genes at the Drosophila wing margin. Dev Dyn 2011; 240: 2051–2059.
Janody F, Martirosyan Z, Benlali A, Treisman JE : Two subunits of the Drosophila mediator complex act together to control cell affinity. Development 2003; 130: 3691–3701.
Stuhlmiller TJ, Garcia-Castro MI : Current perspectives of the signaling pathways directing neural crest induction. Cel Mol Life Sci 2012; 69: 3715–3737.
Calmont A, Ivins S, Van Bueren KL et al: Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 2009; 136: 3173–3183.
Lin AE, Pober BR, Mullen MP, Slavotinek AM : Cardiovascular malformations in Fryns syndrome: is there a pathogenic role for neural crest cells? Am J Med Genet A 2005; 139: 186–193.
Angus SP, Nevins JR : A role for Mediator complex subunit MED13L in Rb/E2F-induced growth arrest. Oncogene 2012; 31: 4709–4717.
Acknowledgements
We sincerely thank the affected individuals and their families for participation and their permission to publish the results. We are grateful to Dr Cédric Le Caignec (Service de Génétique Médicale, Nantes, France) and Dr Björn Menten (Centrum Medische Genetica, Ghent, Belgium) for accepting the citation of their DECIPHER Consortium cases (numbers 255109 and 257915, respectively) in our discussion. We also thank the DECIPHER Consortium. This research was supported by grants from the Swiss National Science Foundation (SNF 320030_135669) and the Forschungskredit of the University of Zurich, grant number 54220201.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Asadollahi, R., Oneda, B., Sheth, F. et al. Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability. Eur J Hum Genet 21, 1100–1104 (2013). https://doi.org/10.1038/ejhg.2013.17
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2013.17
Keywords
This article is cited by
-
Use of whole genome analysis to identify shared genomic variants across breeds in canine mitral valve disease
Human Genetics (2021)
-
Report of a de novo c.2605C > T (p.Pro869Ser) change in the MED13L gene and review of the literature for MED13L-related intellectual disability
Italian Journal of Pediatrics (2020)
-
MED13L-related intellectual disability: involvement of missense variants and delineation of the phenotype
neurogenetics (2018)
-
Regulation of metabolism by the Mediator complex
Biophysics Reports (2016)
-
De novo genic mutations among a Chinese autism spectrum disorder cohort
Nature Communications (2016)

{kind=link}