
Abstract
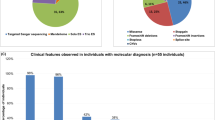
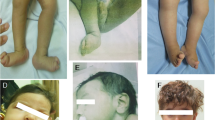
Intellectual disability (ID) is a clinical sign reflecting diverse neurodevelopmental disorders that are genetically and phenotypically heterogeneous. Just recently, partial or complete deletion of methyl-CpG-binding domain 5 (MBD5) gene has been implicated as causative in the phenotype associated with 2q23.1 microdeletion syndrome. In the course of systematic whole-genome screening of individuals with unexplained ID by array-based comparative genomic hybridization, we identified de novo intragenic deletions of MBD5 in three patients leading, as previously documented, to haploinsufficiency of MBD5. In addition, we described a patient with an unreported de novo MBD5 intragenic duplication. Reverse transcriptase-PCR and sequencing analyses showed the presence of numerous aberrant transcripts leading to premature termination codon. To further elucidate the involvement of MBD5 in ID, we sequenced ten coding, five non-coding exons and an evolutionary conserved region in intron 2, in a selected cohort of 78 subjects with a phenotype reminiscent of 2q23.1 microdeletion syndrome. Besides variants most often inherited from an healthy parent, we identified for the first time a de novo nonsense mutation associated with a much more damaging phenotype. Taken together, these results extend the mutation spectrum in MBD5 gene and contribute to refine the associated phenotype of neurodevelopmental disorder.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Laget S, Joulie M, Le Masson F et al: The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE 2010; 5: e11982.
Wagenstaller J, Spranger S, Lorenz-Depiereux B et al: Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet 2007; 81: 768–779.
Jaillard S, Dubourg C, Gerard-Blanluet M et al: 2q23.1 microdeletion identified by array comparative genomic hybridisation: an emerging phenotype with Angelman-like features? J Med Genet 2009; 46: 847–855.
Williams SR, Mullegama SV, Rosenfeld JA et al: Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet 2010; 18: 436–441.
Van Bon BW, Koolen DA, Brueton L et al: The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur J Hum Genet 2010; 18: 163–170.
Chung BH, Stavropoulos J, Marshall CR et al: 2q23 de novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am J Med Genet A 2011; 155: 424–429.
Noh GJ, Graham JM : 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur J Med Genet 2012; 55: 354–357.
Chung BH, Mullegama S, Marshall CR et al: Severe intellectual disability and autistic features associated with microduplication 2q23.1. Eur J Hum Genet 2012; 20: 398–403.
Bonnet C, Masurel-Paulet A, Khan AA et al: Exploring the potential role of disease-causing mutation in a gene desert: duplication of noncoding elements 5′ of GRIA3 is associated with GRIA3 silencing and X-linked intellectual disability. Hum Mutat 2012; 33: 355–358.
Talkowski ME, Mullegama SV, Rosenfeld JA et al: Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 2011; 89: 551–563.
Kleefstra T, Kramer JM, Neveling K et al: Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012; 91: 1–10.
Acknowledgements
We thank the patients and their family for their kind cooperation. We thank the cytogenetics and molecular genetics staff at the Nancy Univesity Hospitals for their expert technical assistance. This study was supported by grants from the French Ministry of Health (DGOS) and the ‘Fondation Jérôme Lejeune’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Bonnet, C., Ali Khan, A., Bresso, E. et al. Extended spectrum of MBD5 mutations in neurodevelopmental disorders. Eur J Hum Genet 21, 1457–1461 (2013). https://doi.org/10.1038/ejhg.2013.22
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2013.22
Keywords
This article is cited by
-
Exploring the genetic etiology of drug-resistant epilepsy: incorporation of exome sequencing into practice
Acta Neurologica Belgica (2022)
-
Composite Sleep Problems Observed Across Smith–Magenis Syndrome, MBD5-Associated Neurodevelopmental Disorder, Pitt–Hopkins Syndrome, and ASD
Journal of Autism and Developmental Disorders (2021)
-
Persistent Lin28 Expression Impairs Neurite Outgrowth and Cognitive Function in the Developing Mouse Neocortex
Molecular Neurobiology (2019)
-
Targeted next generation sequencing: the diagnostic value in early-onset epileptic encephalopathy
Acta Neurologica Belgica (2017)
-
Clinical and Molecular Aspects of MBD5-Associated Neurodevelopmental Disorder (MAND)
European Journal of Human Genetics (2016)
