
Abstract
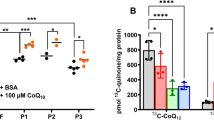
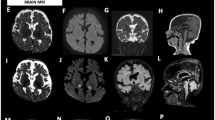
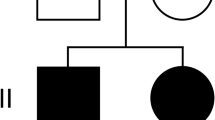
Coenzyme Q10 deficiency is a clinically and genetically heterogeneous disorder, with manifestations that may range from fatal neonatal multisystem failure, to adult-onset encephalopathy. We report a patient who presented at birth with severe lactic acidosis, proteinuria, dicarboxylic aciduria, and hepatic insufficiency. She also had dilation of left ventricle on echocardiography. Her neurological condition rapidly worsened and despite aggressive care she died at 23 h of life. Muscle histology displayed lipid accumulation. Electron microscopy showed markedly swollen mitochondria with fragmented cristae. Respiratory-chain enzymatic assays showed a reduction of combined activities of complex I+III and II+III with normal activities of isolated complexes. The defect was confirmed in fibroblasts, where it could be rescued by supplementing the culture medium with 10 μ M coenzyme Q10. Coenzyme Q10 levels were reduced (28% of controls) in these cells. We performed exome sequencing and focused the analysis on genes involved in coenzyme Q10 biosynthesis. The patient harbored a homozygous c.545T>G, p.(Met182Arg) alteration in COQ2, which was validated by functional complementation in yeast. In this case the biochemical and morphological features were essential to direct the genetic diagnosis. The parents had another pregnancy after the biochemical diagnosis was established, but before the identification of the genetic defect. Because of the potentially high recurrence risk, and given the importance of early CoQ10 supplementation, we decided to treat with CoQ10 the newborn child pending the results of the biochemical assays. Clinicians should consider a similar management in siblings of patients with CoQ10 deficiency without a genetic diagnosis.
Similar content being viewed by others
Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L : Genetics of coenzyme q10 deficiency. Mol Syndromol 2014; 5: 156–162.
Forsgren M, Attersand A, Lake S et al: Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem J 2004; 382: 519–526.
Quinzii CM, Emmanuele V, Hirano M : Clinical presentations of coenzyme q10 deficiency syndrome. Mol Syndromol 2014; 5: 141–146.
Diomedi-Camassei F, Di Giandomenico S, Santorelli FM et al: COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 2007; 18: 2773–2780.
Mollet J, Giurgea I, Schlemmer D et al: Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest 2007; 117: 765–772.
Montini G, Malaventura C, Salviati L : Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 2008; 358: 2849–2850.
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C : Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 2012; 7: 1235–1246.
Montero R, Sanchez-Alcazar JA, Briones P et al: Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin Biochem 2008; 41: 697–700.
Lopez-Martin JM, Salviati L, Trevisson E et al: Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet 2007; 16: 1091–1097.
Andolfo I, Alper SL, De Franceschi L et al: Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 2013; 121: 3925–3935, S3921-3912.
Quinzii C, Naini A, Salviati L et al: A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 2006; 78: 345–349.
Cassandrini D, Cilio MR, Bianchi M et al: Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J Inherit Metab Dis 2013; 36: 43–53.
Trevisson E, Burlina A, Doimo M et al: Functional complementation in yeast allows molecular characterization of missense argininosuccinate lyase mutations. J Biol Chem 2009; 284: 28926–28934.
Salviati L, Sacconi S, Murer L et al: Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 2005; 65: 606–608.
Trevisson E, DiMauro S, Navas P, Salviati L : Coenzyme Q deficiency in muscle. Curr Opin Neurol 2011; 24: 449–456.
Salviati L, Trevisson E, Rodriguez Hernandez MA et al: Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J Med Genet 2012; 49: 187–191.
Emma F, Bertini E, Salviati L, Montini G : Renal involvement in mitochondrial cytopathies. Pediatr Nephrol 2012; 27: 539–550.
Dinwiddie DL, Smith LD, Miller NA et al: Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 2013; 102: 148–156.
Scalais E, Chafai R, Van Coster R et al: Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur J Paediatr Neurol 2013; 17: 625–630.
Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L : Genetic bases and clinical manifestations of coenzyme Q (CoQ) deficiency. J Inherit Metab Dis 2014, e-pub ahead of print 5 August 2014 doi:10.1007/s10545-014-9749-9.
Acknowledgements
This work was supported by grants from Fondazione CARIPARO, Telethon Italy GGP13222 and University of Padova (CPDA123573/12) to LS; Telethon Italy GGP10121 and PRIN 20108WT59Y_003 to OZ; Italian Ministry of Health (GR-2009-1578914) to ET; Spanish FIS grant PI11-00078 and Proyecto Excelencia P08-CTS-03988 to PN.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Desbats, M., Vetro, A., Limongelli, I. et al. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur J Hum Genet 23, 1254–1258 (2015). https://doi.org/10.1038/ejhg.2014.277
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2014.277
This article is cited by
-
Compound heterozygous inheritance of two novel COQ2 variants results in familial coenzyme Q deficiency
Orphanet Journal of Rare Diseases (2020)
-
A steroid-resistant nephrotic syndrome in an infant resulting from a consanguineous marriage with COQ2 and ARSB gene mutations: a case report
BMC Medical Genetics (2019)
-
Vitamin K2 cannot substitute Coenzyme Q10 as electron carrier in the mitochondrial respiratory chain of mammalian cells
Scientific Reports (2019)
-
Treatment of steroid-resistant nephrotic syndrome in the genomic era
Pediatric Nephrology (2019)
-
COQ2 nephropathy: a treatable cause of nephrotic syndrome in children
Pediatric Nephrology (2018)
