
Abstract
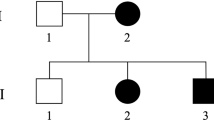
Hereditary cerebellar ataxias and hereditary spastic paraplegias are clinically and genetically heterogeneous and often overlapping neurological disorders. Mutations in SPG7 cause the autosomal recessive spastic paraplegia type 7 (SPG7), but recent studies indicate that they are also one of the most common causes of recessive cerebellar ataxia. In Quebec, a significant number of patients affected with cerebellar ataxia and spasticity remain without a molecular diagnosis. We performed whole-exome sequencing in three French Canadian (FC) patients affected with spastic ataxia and uncovered compound heterozygous variants in SPG7 in all three. Sanger sequencing of SPG7 exons and exon/intron boundaries was used to screen additional patients. In total, we identified recessive variants in SPG7 in 22 FC patients belonging to 12 families (38.7% of the families screened), including two novel variants. The p.(Ala510Val) variant was the most common in our cohort. Cerebellar features, including ataxia, were more pronounced than spasticity in this cohort. These results strongly suggest that variants affecting the function of SPG7 are the fourth most common form of recessive ataxia in FC patients. Thus, we propose that SPG7 mutations explain a significant proportion of FC spastic ataxia cases and that this gene should be considered in unresolved patients.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Noreau A, Dion PA, Rouleau GA : Molecular aspects of hereditary spastic paraplegia. Exp Cell Res 2014; 325: 18–26.
Hersheson J, Haworth A, Houlden H : The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 2012; 33: 1324–1332.
Ruano L, Melo C, Silva MC, Coutinho P : The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014; 42: 174–183.
Casari G, De Fusco M, Ciarmatori S et al: Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998; 93: 973–983.
Wilkinson PA, Crosby AH, Turner C et al: A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain 2004; 127 (Pt 5): 973–980.
van Gassen KL, van der Heijden CD, de Bot ST et al: Genotype-phenotype correlations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain 2012; 135 (Pt 10): 2994–3004.
Klebe S, Depienne C, Gerber S et al: Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012; 135 (Pt 10): 2980–2993.
Kumar KR, Blair NF, Vandebona H et al: Targeted next generation sequencing in SPAST-negative hereditary spastic paraplegia. J Neurol 2013; 260: 2516–2522.
Sanchez-Ferrero E, Coto E, Beetz C et al: SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V. Clin Genet 2013; 83: 257–262.
Arnoldi A, Tonelli A, Crippa F et al: A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat 2008; 29: 522–531.
Doi H, Ohba C, Tsurusaki Y et al: Identification of a novel homozygous SPG7 mutation in a Japanese patient with spastic ataxia: making an efficient diagnosis using exome sequencing for autosomal recessive cerebellar ataxia and spastic paraplegia. Intern Med 2013; 52: 1629–1633.
Orsucci D, Petrucci L, Ienco EC et al: Hereditary spastic paraparesis in adults. A clinical and genetic perspective from Tuscany. Clin Neurol Neurosurg 2014; 120: 14–19.
Racis L, Tessa A, Di Fabio R et al: The high prevalence of hereditary spastic paraplegia in Sardinia, insular Italy. J Neurol 2014; 261: 52–59.
Elleuch N, Depienne C, Benomar A et al: Mutation analysis of the paraplegin gene (SPG7) in patients with hereditary spastic paraplegia. Neurology 2006; 66: 654–659.
Roxburgh RH, Marquis-Nicholson R, Ashton F et al: The p.Ala510Val mutation in the SPG7 (paraplegin) gene is the most common mutation causing adult onset neurogenetic disease in patients of British ancestry. J Neurol 2013; 260: 1286–1294.
Pfeffer G, Pyle A, Griffin H et al: SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 2015; 84: 1174–1176.
van de Warrenburg BP, van Gaalen J, Boesch S et al: EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol 2014; 21: 552–562.
Dupre N, Gros-Louis F, Bouchard JP, Noreau A, Roueau GA . SYNE1-Related autosomal recessive cerebellar ataxia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al (eds): GeneReviews(R). University of Washington: Seattle, WA, USA, 1993.
Yoon G, Baskin B, Tarnopolsky M et al: Autosomal recessive hereditary spastic paraplegia-clinical and genetic characteristics of a well-defined cohort. Neurogenetics 2013; 14: 181–188.
Li H, Handsaker B, Wysoker A et al: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Wang K, Li M, Hakonarson H : ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Bonn F, Pantakani K, Shoukier M, Langer T, Mannan AU : Functional evaluation of paraplegin mutations by a yeast complementation assay. Hum Mutat 2010; 31: 617–621.
Pfeffer G, Gorman GS, Griffin H et al: Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014; 137 (Pt 5): 1323–1336.
Marcotulli C, Leonardi L, Tessa A et al: Early-onset optic neuropathy as initial clinical presentation in SPG7. J Neurol 2014; 261: 1820–1821.
Brugman F, Scheffer H, Wokke JH et al: Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology 2008; 71: 1500–1505.
Thiffault I, Dicaire MJ, Tetreault M et al: Diversity of ARSACS mutations in French-Canadians. Can J Neurol Sci 2013; 40: 61–66.
Acknowledgements
We thank the patients and their relatives who accepted to partake in this study. This project was financially supported by the Fondation Monaco. This work was selected for study by the Care4Rare (Enhanced Care for Rare Genetic Diseases in Canada) Consortium Gene Discovery Steering Committee: Kym Boycott (lead; University of Ottawa), Alex MacKenzie (co-lead; University of Ottawa), Jacek Majewski (McGill University), Michael Brudno (University of Toronto), Dennis Bulman (University of Ottawa), and David Dyment (University of Ottawa) and was funded in part by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec and the Children’s Hospital of Eastern Ontario Foundation. The authors wish to acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Génome Québec Innovation Centre, Montréal, Canada. KC and RLP received a Doctoral Award from the Fonds de recherche du Québec - Santé (FRQS). MT received a post-doctoral award from the Réseau de Médecine Génétique Appliquée and FRQS.
Web resources
EVS: https://evs.gs.washington.edu/EVS/. ExAC: Cambridge, MA, USA (http://exac.broadinstitute.org; March 2015). MutationTaster: www.mutationtaster.org
Accession codes
Gene: SPG7. Variants/accession numbers: c.1529C>T, p.(Ala510Val)/SCV000245719; c.2249C>T, p.(Pro750Leu)/SCV000245720; c.1715C>T, p.(Ala572Val)/SCV000245721; c.988-1G>A/SCV000245722; c.1045G>A, p.(Gly349Ser)/SCV000245723; c.233T>A, p.(Leu78Ter)/SCV000245724; c.473_474del, p.(Leu158QfsTer30)/SCV000245726.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Choquet, K., Tétreault, M., Yang, S. et al. SPG7 mutations explain a significant proportion of French Canadian spastic ataxia cases. Eur J Hum Genet 24, 1016–1021 (2016). https://doi.org/10.1038/ejhg.2015.240
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2015.240
This article is cited by
-
An integrated modelling methodology for estimating global incidence and prevalence of hereditary spastic paraplegia subtypes SPG4, SPG7, SPG11, and SPG15
BMC Neurology (2022)
-
Autosomal recessive adult onset ataxia
Journal of Neurology (2022)
-
Positive DAT-SCAN in SPG7: a case report mimicking possible MSA-C
BMC Neurology (2021)
-
Molecular epidemiology of hereditary ataxia in Finland
BMC Neurology (2021)
-
Inherited Cerebellar Ataxias: 5-Year Experience of the Irish National Ataxia Clinic
The Cerebellum (2021)
