
Abstract
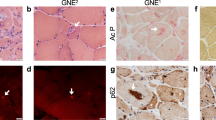
GNE myopathy is an autosomal recessive adult-onset disorder characterized by progressive muscle atrophy and weakness, initially involving the distal muscles, while often sparing the quadriceps. It is caused by variants in the GNE gene that encodes a key bifunctional enzyme in the sialic acid biosynthetic pathway. We investigated the clinical and molecular characteristics of 18 non-Jewish Persian patients from 11 unrelated GNE myopathy families. In addition, we reviewed the previously reported cases and suggest genotype–phenotype correlations for the identified variants. Comprehensive clinical and laboratory evaluations were carried out. Sequencing of the GNE gene was performed using genomic DNA from the patients. Screening of the identified variants was performed in all relevant family members. Molecular analyses identified three causative homozygous GNE variants in 11 families: c.2228T>C (p. M743T) in 7, c.830G>A (p.R277Q) in 2, and one novel variation (c.804G>A) in 2 families that results in a synonymous codon change (p.L268=) and likely creates a novel splice site affecting the protein function. This study confirms that c.2228T>C (p.M743T) is the most prevalent disease-causing variant in the non-Jewish Persian population, but other GNE variants can cause GNE myopathy in this population. The patients with all three different variants had similar ages of onset. The youngest patient was an 18-year-old girl in whom the c.830G>A (p.R277Q) variant was identified, whereas the oldest onset age (31 years) was seen in a male patient with c.804G>A (p.L268=). The results of this investigation expand our knowledge about the genotype–phenotype correlations in GNE myopathy and aid in clinical management and therapeutic interventions.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Argov Z, Yarom R : "Rimmed vacuole myopathy" sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci 1984; 64: 33–43.
Sadeh M, Gadoth N, Hadar H, Ben-David E : Vacuolar myopathy sparing the quadriceps. Brain 1993; 116 (Pt 1): 217–232.
Nonaka I, Noguchi S, Nishino I : Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep 2005; 5: 61–65.
Krause S, Schlotter-Weigel B, Walter MC et al: A novel homozygous missense mutation in the GNE gene of a patient with quadriceps-sparing hereditary inclusion body myopathy associated with muscle inflammation. Neuromuscul Disord 2003; 13: 830–834.
Argov Z, Eisenberg I, Grabov-Nardini G et al: Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology 2003; 60: 1519–1523.
Lu X, Pu C, Huang X, Liu J, Mao Y : Distal myopathy with rimmed vacuoles: clinical and muscle morphological characteristics and spectrum of GNE gene mutations in 53 Chinese patients. Neurol Res 2011; 33: 1025–1031.
Eisenberg I, Avidan N, Potikha T et al: The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 2001; 29: 83–87.
Kayashima T, Matsuo H, Satoh A et al: Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene (GNE). J Hum Genet 2002; 47: 77–79.
Hinderlich S, Stasche R, Zeitler R, Reutter W : A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem 1997; 272: 24313–24318.
Stasche R, Hinderlich S, Weise C et al: A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Molecular cloning and functional expression of UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem 1997; 272: 24319–24324.
Kelm S, Schauer R : Sialic acids in molecular and cellular interactions. Int Rev Cytol 1997; 175: 137–240.
Ulloa-Aguirre A, Timossi C, Damian-Matsumura P, Dias JA : Role of glycosylation in function of follicle-stimulating hormone. Endocrine 1999; 11: 205–215.
Horstkorte R, Nohring S, Wiechens N et al: Tissue expression and amino acid sequence of murine UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase. Eur J Biochem 1999; 260: 923–927.
Schwarzkopf M, Knobeloch KP, Rohde E et al: Sialylation is essential for early development in mice. Proc Natl Acad Sci USA 2002; 99: 5267–5270.
Weidemann W, Stelzl U, Lisewski U et al: The collapsin response mediator protein 1 (CRMP-1) and the promyelocytic leukemia zinc finger protein (PLZF) bind to UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE), the key enzyme of sialic acid biosynthesis. FEBS Lett 2006; 580: 6649–6654.
Huizing M, Rakocevic G, Sparks SE et al: Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab 2004; 81: 196–202.
Ricci E, Broccolini A, Gidaro T et al: NCAM is hyposialylated in hereditary inclusion body myopathy due to GNE mutations. Neurology 2006; 66: 755–758.
Hinderlich S, Salama I, Eisenberg I et al: The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered overall cellular sialylation in hereditary inclusion body myopathy. FEBS Lett 2004; 566: 105–109.
Broccolini A, Gidaro T, De Cristofaro R et al: Hyposialylation of neprilysin possibly affects its expression and enzymatic activity in hereditary inclusion-body myopathy muscle. J Neurochem 2008; 105: 971–981.
Tajima Y, Uyama E, Go S et al: Distal myopathy with rimmed vacuoles: impaired O-glycan formation in muscular glycoproteins. Am J Pathol 2005; 166: 1121–1130.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C : Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
Darvish D, Vahedifar P, Huo Y : Four novel mutations associated with autosomal recessive inclusion body myopathy (MIM: 600737). Mol Genet Metab 2002; 77: 252–256.
Askanas V, Engel WK : Muscle Aging, Inclusion-Body Myositis and Myopathies. Oxford, UK Wiley-Blackwell, 2012.
Nishino I, Noguchi S, Murayama K et al: Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology 2002; 59: 1689–1693.
Nalini A, Gayathri N, Dawn R : Distal myopathy with rimmed vacuoles: report on clinical characteristics in 23 cases. Neurol India 2010; 58: 235–241.
Khademian H, Mehravar E, Urtizberea J et al: Prevalence of GNE p.M712T and hereditary inclusion body myopathy (HIBM) in Sangesar population of Northern Iran. Clin Genet 2013; 84: 589–592.
Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG : Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet 1996; 5: 159–163.
Malicdan MC, Noguchi S, Nishino I : Perspectives on distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy: contributions from an animal model. Lack of sialic acid, a central determinant in sugar chains, causes myopathy? Acta Myol 2007; 26: 171–175.
Park YE, Kim HS, Choi ES et al: Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutations. J Neurol Sci 2012; 321: 77–81.
Huizing M, Krasnewich DM : Hereditary inclusion body myopathy: a decade of progress. Biochim Biophys Acta 2009; 1792: 881–887.
Hinderlich S, Weidemann W, Yardeni T, Horstkorte R, Huizing M : UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE): a master regulator of sialic acid synthesis. Top Curr Chem 2013, e-pub ahead of print 11 July 2013.
Amouri R, Driss A, Murayama K, Kefi M, Nishino I, Hentati F : Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscul Disord 2005; 15: 361–363.
Sim JE, Park HJ, Shin HY, Nam TS, Kim SM, Choi YC : Clinical characteristics and molecular genetic analysis of Korean patients with GNE myopathy. Yonsei Med J 2013; 54: 578–582.
Milman Krentsis I, Sela I, Eiges R et al: GNE is involved in the early development of skeletal and cardiac muscle. PLoS One 2011; 6: e21389.
Sivakumar K, Dalakas MC : The spectrum of familial inclusion body myopathies in 13 families and a description of a quadriceps-sparing phenotype in non-Iranian Jews. Neurology 1996; 47: 977–984.
Broccolini A, Pescatori M, D'Amico A et al: An Italian family with autosomal recessive inclusion-body myopathy and mutations in the GNE gene. Neurology 2002; 59: 1808–1809.
Diaz A, Montfort M, Cormand B et al: Gaucher disease: the N370S mutation in Ashkenazi Jewish and Spanish patients has a common origin and arose several thousand years ago. Am J Hum Genet 1999; 64: 1233–1238.
Eisenberg I, Grabov-Nardini G, Hochner H et al: Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat 2003; 21: 99.
Grandis M, Gulli R, Cassandrini D et al: The spectrum of GNE mutations: allelic heterogeneity for a common phenotype. Neurol Sci 2010; 31: 377–380.
Cho A, Hayashi YK, Monma K et al: Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy). J Neurol Neurosurg Psychiatry 2013; 85: 914–917.
Mori-Yoshimura M, Monma K, Suzuki N et al: Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. J Neurol Sci 2012; 318: 100–105.
Tomimitsu H, Shimizu J, Ishikawa K, Ohkoshi N, Kanazawa I, Mizusawa H : Distal myopathy with rimmed vacuoles (DMRV): new GNE mutations and splice variant. Neurology 2004; 62: 1607–1610.
Sparks SE, Ciccone C, Lalor M et al: Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human hereditary inclusion body myopathy. Glycobiology 2005; 15: 1102–1110.
Saechao C, Valles-Ayoub Y, Esfandiarifard S et al: Novel GNE mutations in hereditary inclusion body myopathy patients of non-Middle Eastern descent. Genet Test Mol Biomarkers 2010; 14: 157–162.
Stober A, Aleo A, Kuhl V et al: Novel missense mutation p.A310P in the GNE gene in autosomal-recessive hereditary inclusion-body myopathy/distal myopathy with rimmed vacuoles in an Italian family. Neuromuscul Disord 2010; 20: 335–336.
Chu CC, Kuo HC, Yeh TH, Ro LS, Chen SR, Huang CC : Heterozygous mutations affecting the epimerase domain of the GNE gene causing distal myopathy with rimmed vacuoles in a Taiwanese family. Clin Neurol Neurosurg 2007; 109: 250–256.
Broccolini A, Ricci E, Cassandrini D et al: Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion-body myopathy. Hum Mutat 2004; 23: 632.
Li H, Chen Q, Liu F et al: Clinical and molecular genetic analysis in Chinese patients with distal myopathy with rimmed vacuoles. J Hum Genet 2011; 56: 335–338.
Niethamer TK, Yardeni T, Leoyklang P et al: Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab 2012; 107: 748–755.
Malicdan MC, Noguchi S, Hayashi YK, Nonaka I, Nishino I : Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med 2009; 15: 690–695.
Acknowledgements
We would like to thank Dr Hamidreza Haghighi for his useful comments. This study was undertaken as part of GENE-ME (a research initiative for investigation of inherited diseases from the Middle East).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Haghighi, A., Nafissi, S., Qurashi, A. et al. Genetics of GNE myopathy in the non-Jewish Persian population. Eur J Hum Genet 24, 243–251 (2016). https://doi.org/10.1038/ejhg.2015.78
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2015.78
This article is cited by
-
Generation and Characterization of a Skeletal Muscle Cell-Based Model Carrying One Single Gne Allele: Implications in Actin Dynamics
Molecular Neurobiology (2021)
-
Quantitative nuclear magnetic resonance imaging detects subclinical changes over 1 year in skeletal muscle of GNE myopathy
Journal of Neurology (2020)
-
GNE myopathy: from clinics and genetics to pathology and research strategies
Orphanet Journal of Rare Diseases (2018)
