
Abstract
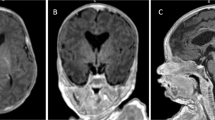
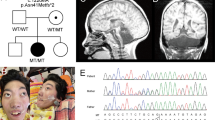
We report an 8-year-old boy with a complex cerebral malformation, intellectual disability, and complex partial seizures. Whole-exome sequencing revealed a yet unreported de novo variant in the PIK3R2 gene that was recently associated with megalencephaly–polymicrogyria–polydactyly–hydrocephalus (MPPH) syndrome and bilateral perisylvian polymicrogyria (BPP). Our patient showed cerebral abnormalities (megalencephaly, perisylvian polymicrogyria, and mega corpus callosum) that were consistent with these conditions. Imaging also showed right temporal anomalies suggestive of cortical dysplasia. Until now, only three variants (c.1117G>A (p.(G373R)), c.1126A>G (p.(K376E)) and c.1202T>C (p.(L401P))) affecting the SH2 domain of the PIK3R2 protein have been reported in MPPH and BPP syndromes. In contrast to the variants reported so far, the patient described herein exhibits the c.1669G>C (p.(D557H)) variant that affects a highly conserved residue at the interface with the PI3K catalytic subunit α. The phenotypic spectrum associated with variants in this gene and its pathway are likely to continue to expand as more cases are identified.
Similar content being viewed by others
Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Mirzaa GM, Riviere JB, Dobyns WB : Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet 2013; 163C: 122–130.
Riviere JB, Mirzaa GM, O'Roak BJ et al: De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 2012; 44: 934–940.
Keppler-Noreuil KM, Rios JJ, Parker VE et al: PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A 2015; 167A: 287–295.
Lee JH, Huynh M, Silhavy JL et al: De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012; 44: 941–945.
Mirzaa GM, Conti V, Timms AE et al: Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: a next-generation sequencing study. Lancet Neurol 2015; 14: 1182–1195.
Mirzaa GM, Parry DA, Fry AE et al: De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet 2014; 46: 510–515.
Luks VL, Kamitaki N, Vivero MP et al: Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr 2015; 166: 1048–1054, e1041-1045.
Alfaiz AA, Micale L, Mandriani B et al: TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat 2014; 35: 447–451.
Borck G, Hog F, Dentici ML et al: BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res 2015; 25: 609.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Nakamura K, Kato M, Tohyama J et al: AKT3 and PIK3R2 mutations in two patients with megalencephaly-related syndromes: MCAP and MPPH. Clin Genet 2014; 85: 396–398.
Tapper WJ, Foulds N, Cross NC et al: Megalencephaly syndromes: exome pipeline strategies for detecting low-level mosaic mutations. PLoS ONE 2014; 9: e86940.
Mirzaa GM, Conway RL, Gripp KW et al: Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A 2012; 158A: 269–291.
Blumcke I, Thom M, Aronica E et al: The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011; 52: 158–174.
Jansen LA, Mirzaa GM, Ishak GE et al: PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015; 138: 1613–1628.
Lim JS, Kim WI, Kang HC et al: Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015; 21: 395–400.
Shain C, Ramgopal S, Fallil Z et al: Polymicrogyria-associated epilepsy: a multicenter phenotypic study from the Epilepsy Phenome/Genome Project. Epilepsia 2013; 54: 1368–1375.
Acknowledgements
Authors thank the family for its contribution and the members of the Lausanne Genomic Technologies Facility. AAA is recipient of a scholarship from the Saudi Arabian National Guard Health Affairs. This work was supported by a grant of the Swiss National Science Foundation 31003A_160203 and the Lithuanian-Swiss Cooperation Programme (AR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Note added in proof
During the revision of this manuscript, Mirzaa et al.5 reported PIK3R2 variants in BPP, we modified our manuscript to discuss our results in view of these recently published data to help potential readers getting a complete view of current knowledge.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Terrone, G., Voisin, N., Abdullah Alfaiz, A. et al. De novo PIK3R2 variant causes polymicrogyria, corpus callosum hyperplasia and focal cortical dysplasia. Eur J Hum Genet 24, 1359–1362 (2016). https://doi.org/10.1038/ejhg.2016.7
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2016.7
