
Abstract
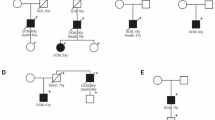
Dilated cardiomyopathy (DCM) is extremely heterogeneous with a large proportion due to dominantly inherited disease-causing variants in sarcomeric genes. Recessive metabolic diseases may cause DCM, usually with onset in childhood, and in the context of systemic disease. Whether metabolic defects can also cause adult-onset DCM is currently unknown. Therefore, we performed an extensive metabolic screening in 36 consecutive adult-onset DCM patients. Diagnoses were confirmed by Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA). Measurement of propionyl-CoA carboxylase (PCC) activity was done in fibroblasts. Whole exome sequencing (WES) data of 157 additional DCM patients were analyzed for genetic defects. We found a metabolic profile characteristic for propionic acidemia in a patient with severe DCM from 55 years of age. Genetic analysis demonstrated compound heterozygous variants in PCCA. Enzymatic activity of PCC in fibroblasts was markedly reduced. A targeted analysis of the PCCA and PCCB genes using available WES data from 157 further DCM patients subsequently identified another patient with propionic acidemia. This patient had compound heterozygous variants in PCCB, and developed severe DCM from 42 years of age. Adult-onset DCM can be caused by propionic acidemia, an autosomal recessive inheritable metabolic disorder usually presenting as neonatal or childhood disease. Current guidelines advise a low-protein diet to ameliorate or prevent detrimental aspects of the disease. Long-term follow-up of a larger group of patients may show whether this diet would also ameliorate DCM. Our results suggest that diagnostic metabolic screening to identify propionic acidemia and related disorders in DCM patients is justified.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Maron BJ, Towbin JA, Thiene G et al: Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816.
Caforio AL, Pankuweit S, Arbustini E et al: Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34: 2636–2648, 2648a–2648d.
Hershberger RE, Hedges DJ, Morales A : Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013; 10: 531–547.
Hazebroek MR, Moors S, Dennert R et al: Prognostic relevance of gene–environment interactions in patients with dilated cardiomyopathy: applying the MOGE(S) classification. J Am Coll Cardiol 2015; 66: 1313–1323.
Michels VV, Moll PP, Miller FA et al: The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992; 326: 77–82.
Grünig E, Tasman JA, Kücherer H, Franz W, Kübler W, Katus HA : Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 1998; 31: 186–194.
Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ : Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol 1998; 31: 195–201.
Posafalvi A, Herkert JC, Sinke RJ et al: Clinical utility gene card for: dilated cardiomyopathy (CMD). Eur J Hum Genet 2013; 21, doi:10.1038/ejhg.2012.276.
Mestroni L, Rocco C, Gregori D et al: Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol 1999; 34: 181–190.
D'Adamo P, Fassone L, Gedeon A et al: The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61: 862–867.
Levitas A, Muhammad E, Harel G et al: Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet 2010; 18: 1160–1165.
Lefeber DJ, de Brouwer AP, Morava E et al: Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet 2011; 7: e1002427.
Müller T, Krasnianski M, Witthaut R, Deschauer M, Zierz S : Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul Disord 2005; 15: 372–376.
Gottdiener JS, Bednarz J, Devereux R et al: American Society of Echocardiography recommendations for use of echocardiography in clinical trials. J Am Soc Echocardiogr 2004; 17: 1086–1119.
Heymans S, Schroen B, Vermeersch P et al: Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 2005; 112: 1136–1144.
Dennert R, Velthuis S, Schalla S et al: Intravenous immunoglobulin therapy for patients with idiopathic cardiomyopathy and endomyocardial biopsy-proven high PVB19 viral load. Antivir Ther 2010; 15: 193–201.
Bock CT, Klingel K, Kandolf R : Human parvovirus B19-associated myocarditis. N Engl J Med 2010; 362: 1248–1249.
Richardson P, McKenna W, Bristow M et al: Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93: 841–842.
Waterval WAH, Scheijen JLJM, Ortmans-Ploemen MMJC, Habets-van der Poel CD, Bierau J : Quantitative UPLC-MS/MS analysis of underivatised amino acids in body fluids is a reliable tool for the diagnosis and follow-up of patients with inborn errors of metabolism. Clin Chim Acta 2009; 407: 36–42.
Vreken P, van Lint AE, Bootsma AH, Overmars H, Wanders RJ, van Gennip AH : Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects. J Inherit Metab Dis 1999; 22: 302–306.
Blau N, Duran M, Gibson KM : Laboratory Guide to the Methods in Biochemical Genetics. Berlin Heidelberg: Springer-Verlag, 2008.
de Jong G, van Noort WL, van Eijk HG : Optimized separation and quantitation of serum and cerebrospinal fluid transferrin subfractions defined by differences in iron saturation or glycan composition. Adv Exp Med Biol 1994; 356: 51–59.
O'Roak BJ, Vives L, Fu W et al: Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012; 338: 1619–1622.
Baumgartner MR, Horster F, Dionisi-Vici C et al: Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014; 9: 130.
Hall PR, Wang YF, Rivera-Hainaj RE et al: Transcarboxylase 12S crystal structure: hexamer assembly and substrate binding to a multienzyme core. EMBO J 2003; 22: 2334–2347.
Lek M, Karczewski KJ, Minikel EV et al: Analysis of protein-coding genetic variation in 60 706 humans. Nature 2016; 536: 285–291.
Fragaki K, Cano A, Benoist JF et al: Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion 2011; 11: 533–536.
Baruteau J, Hargreaves I, Krywawych S et al: Successful reversal of propionic acidaemia associated cardiomyopathy: evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion 2014; 17: 150–156.
Shchelochkov OA, Carrillo N, Venditti C . Propionic Acidemia; 2012 May 17 [Updated 2016 Oct 6]; In: Pagon RA, Adam MP, Ardinger HH et al. (eds): GeneReviews® [Internet]. University of Washington: Seattle, WA, 1993–2017.
Massoud AF, Leonard JV : Cardiomyopathy in propionic acidaemia. Eur J Pediatr 1993; 152: 441–445.
Romano S, Valayannopoulos V, Touati G et al: Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J Pediatr 2010; 156: 128–134.
Lee TM, Addonizio LJ, Barshop BA, Chung WK : Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J Inherit Metab Dis 2009; 32 (Suppl 1): S97–S101.
Laemmle A, Balmer C, Doell C, Sass JO, Haberle J, Baumgartner MR : Propionic acidemia in a previously healthy adolescent with acute onset of dilated cardiomyopathy. Eur J Pediatr 2014; 173: 971–974.
Kraus JP, Spector E, Venezia S et al: Mutation analysis in 54 propionic acidemia patients. J Inherit Metab Dis 2012; 35: 51–63.
den Hollander AI, Koenekoop RK, Yzer S et al: Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 2006; 79: 556–561.
Costes B, Girodon E, Ghanem N et al: Frequent occurrence of the CFTR intron 8 (TG)n 5T allele in men with congenital bilateral absence of the vas deferens. Eur J Hum Genet 1995; 3: 285–293.
Drenth JP, Cuisset L, Grateau G et al: Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22: 178–181.
Guenzel AJ, Hofherr SE, Hillestad M et al: Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol Ther 2013; 21: 1316–1323.
Hofherr SE, Senac JS, Chen CY, Palmer DJ, Ng P, Barry MA : Short-term rescue of neonatal lethality in a mouse model of propionic acidemia by gene therapy. Hum Gene Ther 2009; 20: 169–180.
Acknowledgements
We would like to thank Irene Körver-Keularts, PhD and Daphna Habets, PhD from the Department of Clinical Genetics, Maastricht University Medical Center+, the Netherlands for assistance with the interpretation of the metabolic screening, Dirk Lefeber, PhD from the Department of Neurology and Translation Metabolic Laboratory, Radboudumc Nijmegen, the Netherlands for performing the mucopolysaccharides analysis and Christian Gilissen, PhD from the Department of Human Genetics, Radboudumc Nijmegen, the Netherlands for providing the Gene Checker tool. Warsha Kanhai and Matilde Fernandez Ojeda are acknowledged for their enthusiastic and excellent laboratory assistance in the Metabolic Unit, Amsterdam, the Netherlands.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Riemersma, M., Hazebroek, M., Helderman-van den Enden, A. et al. Propionic acidemia as a cause of adult-onset dilated cardiomyopathy. Eur J Hum Genet 25, 1195–1201 (2017). https://doi.org/10.1038/ejhg.2017.127
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2017.127
This article is cited by
-
Sarcomeric remodelling in human heart failure unraveled by single molecule long read sequencing
EMBO Molecular Medicine (2026)
-
Metabolomic and transcriptomic insights into the mechanisms of renal ischemia-reperfusion injury progression
Scientific Reports (2024)
-
Therapeutic potential of living donor liver transplantation from heterozygous carrier donors in children with propionic acidemia
Orphanet Journal of Rare Diseases (2022)
-
Methylmalonic and propionic acidemia among hospitalized pediatric patients: a nationwide report
Orphanet Journal of Rare Diseases (2019)
-
Einthoven dissertation prizes 2017
Netherlands Heart Journal (2018)

{kind=link}
{kind=link}