
Abstract
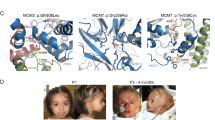
Meier-Gorlin syndrome (MGORS) is a rare disorder characterized by primordial dwarfism, microtia, and patellar aplasia/hypoplasia. Recessive mutations in ORC1, ORC4, ORC6, CDT1, CDC6, and CDC45, encoding members of the pre-replication (pre-RC) and pre-initiation (pre-IC) complexes, and heterozygous mutations in GMNN, a regulator of cell-cycle progression and DNA replication, have already been associated with this condition. We performed whole-exome sequencing (WES) in a patient with a clinical diagnosis of MGORS and identified biallelic variants in MCM5. This gene encodes a subunit of the replicative helicase complex, which represents a component of the pre-RC. Both variants, a missense substitution within a conserved domain critical for the helicase activity, and a single base deletion causing a frameshift and a premature stop codon, were predicted to be detrimental for the MCM5 function. Although variants of MCM5 have never been reported in specific human diseases, defect of this gene in zebrafish causes a phenotype of growth restriction overlapping the one associated with orc1 depletion. Complementation experiments in yeast showed that the plasmid carrying the missense variant was unable to rescue the lethal phenotype caused by mcm5 deletion. Moreover cell-cycle progression was delayed in patient’s cells, as already shown for mutations in the ORC1 gene. Altogether our findings support the role of MCM5 as a novel gene involved in MGORS, further emphasizing that this condition is caused by impaired DNA replication.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
de Munnik SA, Hoefsloot EH, Roukema J et al: Meier-Gorlin syndrome. Orphanet J Rare Dis 2015; 10: 114.
Bicknell LS, Walker S, Klingseisen A et al: Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet 2011; 43: 350–355.
Guernsey DL, Matsuoka M, Jiang H et al: Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat Genet 2011; 43: 360–364.
de Munnik SA, Bicknell LS, Aftimos S et al: Meier-Gorlin syndrome genotype-phenotype studies: 35 individuals with pre-replication complex gene mutations and 10 without molecular diagnosis. Eur J Hum Genet 2012; 20: 598–606.
Burrage LC, Charng WL, Eldomery MK et al: De Novo GMNN mutations cause autosomal-dominant primordial dwarfism associated with Meier-Gorlin syndrome. Am J Hum Genet 2015; 97: 904–913.
Fenwick AL, Kliszczak M, Cooper F et al: Mutations in CDC45, encoding an essential component of the pre-initiation complex, cause meier-gorlin syndrome and craniosynostosis. Am J Hum Genet 2016; 99: 125–138.
Vetro A, Iascone M, Limongelli I et al: Loss-of-function FANCL mutations associate with severe fanconi anemia overlapping the VACTERL association. Hum Mutat 2015; 36: 562–568.
Vetro A, Manolakos E, Petersen MB et al: Unexpected results in the constitution of small supernumerary marker chromosomes. Eur J Med Genet 2012; 55: 185–190.
German J, Schonberg S, Louie E, Chaganti RS : Bloom's syndrome. IV. Sister-chromatid exchanges in lymphocytes. Am J Hum Genet 1977; 29: 248–255.
Thorvaldsdottir H, Robinson JT, Mesirov JP : Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–192.
Ramey CJ, Sclafani RA : Functional conservation of the pre-sensor one beta-finger hairpin (PS1-hp) structures in mini-chromosome maintenance proteins of Saccharomyces cerevisiae and archaea. G3 (Bethesda) 2014; 4: 1319–1326.
Bicknell LS, Bongers EMHF, Leitch A et al: Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet 2011; 43: 356–359.
Ryu S, Holzschuh J, Erhardt S, Ettl AK, Driever W : Depletion of minichromosome maintenance protein 5 in the zebrafish retina causes cell-cycle defect and apoptosis. Proc Natl Acad Sci USA 2005; 102: 18467–18472.
Brewster AS, Wang G, Yu X et al: Crystal structure of a near-full-length archaeal MCM: functional insights for an AAA+ hexameric helicase. Proc Natl Acad Sci USA 2008; 105: 20191–20196.
McGeoch AT, Trakselis MA, Laskey RA, Bell SD : Organization of the archaeal MCM complex on DNA and implications for the helicase mechanism. Nat Struct Mol Biol 2005; 12: 756–762.
Lam SK, Ma X, Sing TL, Shilton BH, Brandl CJ, Davey MJ : The PS1 hairpin of Mcm3 is essential for viability and for DNA unwinding in vitro. PLoS One 2013; 8: e82177.
Trevisson E, Burlina A, Doimo M et al: Functional complementation in yeast allows molecular characterization of missense argininosuccinate lyase mutations. J Biol Chem 2009; 284: 28926–28934.
Gineau L, Cognet C, Kara N et al: Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest 2012; 122: 821–832.
Hughes CR, Guasti L, Meimaridou E et al: MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest 2012; 122: 814–820.
Mirzaa GM, Vitre B, Carpenter G et al: Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum Genet 2014; 133: 1023–1039.
Rauch A, Thiel CT, Schindler D et al: Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 2008; 319: 816–819.
Hossain M, Stillman B : Meier-Gorlin syndrome mutations disrupt an Orc1 CDK inhibitory domain and cause centrosome reduplication. Genes Dev 2012; 26: 1797–1810.
Ge XQ, Jackson DA, Blow JJ : Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 2007; 21: 3331–3341.
Acknowledgements
AV benefits of a research position granted by the University of Pavia in the context of the strategic plan: Towards a governance model for international migration: an interdisciplinary and diachronic perspective (MIGRAT-IN-G). LS is supported by Telethon Italy Grant GGP13222, a grant from Fondazione CARIPARO, and University of Padova Grant CPDA123573/12; OZ is supported by Telethon Italy Grant GGP13060. The URLs for data presented herein are as follows: Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, release ESP6500SI-V2 (http://evs.gs.washington.edu/EVS); ExAC (http://exac.broadinstitute.org); Integrative Genomics Viewer (IGV),http://www.broadinstitute.org/igv/Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/ Phyre2 webserver http://www.sbg.bio.ic.ac.uk/phyre2/ Protein Data Bank (PDB), http://www.rcsb.org/pdb/home/home.do; and RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Vetro, A., Savasta, S., Russo Raucci, A. et al. MCM5: a new actor in the link between DNA replication and Meier-Gorlin syndrome. Eur J Hum Genet 25, 646–650 (2017). https://doi.org/10.1038/ejhg.2017.5
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2017.5
This article is cited by
-
Mcm5 mutation leads to silencing of Stat1-bcl2 which accelerating apoptosis of immature T lymphocytes with DNA damage
Cell Death & Disease (2025)
-
The genetic basis of human height
Nature Reviews Genetics (2025)
-
Site directed mutagenesis reveals functional importance of conserved amino acid residues within the N-terminal domain of Dpb2 in budding yeast
Archives of Microbiology (2025)
-
Differentiated muscle cells of salamander Pleurodeles waltl re-enter the cell cycle
Histochemistry and Cell Biology (2025)
-
A novel homozygous intronic variant in CDT1 that alters splicing causes Meier–Gorlin syndrome, and a review of published mutations and growth hormone treatments
Orphanet Journal of Rare Diseases (2024)
