
Abstract
Purpose:
Fragile X syndrome, a common cause of intellectual disability, is usually caused by CGG trinucleotide expansion in the FMR1 gene. CGG repeat size correlates with expansion risk. Premutation alleles (55–200 repeats) may expand to full mutations in female meiosis. Interspersed AGG repeats decrease allele instability and expansion risk. The carrier rate and stability of FMR1 alleles were evaluated in large cohorts of Ashkenazi and non-Ashkenazi women.
Methods:
A total of 4,344 Ashkenazi and 4,985 non-Ashkenazi cases were analyzed using Southern blotting and polymerase chain reaction between 2004 and 2011. In addition, AGG interruptions were evaluated in 326 Ashkenazi and 298 non-Ashkenazi women who were recruited during 2011.
Results:
Both groups had major peaks of 30 and 29 repeats. Ashkenazi women had a higher frequency of 30 repeats and a lower frequency of other peaks (P < 0.0001). A higher rate of premutations in the 55–59 repeats range (1:114 vs. 1:277) was detected among the Ashkenazi women. Loss of AGG interruptions (<2) was significantly less common among Ashkenazi women (9 vs. 19.5% for non-Ashkenazi women, P = 0.0002).
Conclusion:
Ashkenazi women have a high fragile X syndrome carrier rate and mostly lower-range premutations, and carry a low risk for expansion to a full mutation. Normal-sized alleles in Ashkenazi women have higher average number of AGG interruptions that may increase stability. These factors may decrease the risk for fragile X syndrome offspring among Ashkenazi women.
Genet Med 16 12, 940–944.
Similar content being viewed by others

Introduction
Fragile X syndrome (FXS; OMIM 300624) is the most common form of inherited mental impairment, with a prevalence of approximately 1/4,500 in males. The syndrome is characterized by moderate intellectual disability and behavioral abnormalities, including autistic-like behavior.1,2 The vast majority of FXS cases are caused by an expansion of CGG nucleic-base repeats in the Fragile X mental retardation gene (FMR1). Normal individuals usually have ~30 CGG repeats. An expansion to greater than 200 repeats is associated with abnormal methylation and suppression of FMR1 transcription, causing the absence of the gene product, the FMR protein.3 The lack of FMR protein, an RNA-binding protein, is responsible for the syndrome.4
There are four allelic forms of the FMR1 gene with respect to the repeat length. The normal allele form has approximately 5–44 CGG repeats and it is stable on transmission. In these normal alleles, the CGG repeat region is usually interrupted by AGG triplets after every 9 or 10 CGG repeats. These AGG interruptions are believed to maintain repeat integrity by preventing DNA slippage during replication, therefore increasing the repeats’ stability.5 The full-mutation allele form has more than 200 repeats, with several hundred to several thousand repeats being typical. In between those two forms fall the intermediate allele form and the premutation allele form. The intermediate allele form falls between the normal and premutation size ranges (45–54 repeats). A proportion of these alleles are unstable and may expand by a few repeats during meiosis. In this range, expansion to the full-mutation allele may occur over two or more generations but not in a single transmission.6 The premutation allele form is about 55–200 repeats long. It is unstable and commonly expands during transmission. Individuals with the premutations are defined as carriers of the disease. Expansion of an abnormal allele usually occurs when it is transmitted from the mother but not from the father.7 However, although alleles of 55–59 repeats are defined as premutations, only a very few cases of expansion to a full mutation have actually been reported, emphasizing the fact that such allele sizes are generally stable.8
In general, the risk of expansion of a premutation to a full mutation upon transmission to the offspring is associated with three factors. First, it correlates with the number of CGG repeats.9 Second, the number of AGG interruptions also affects stability on transmission, such that alleles with a high number of CGG repeats that lack AGG interruptions are more prone to expansion.5,10 A third factor affecting risk of transmission is the genetic background because some haplotypes are associated with an increased instability as compared with others.11
To date, FXS has been identified in all the ethnic groups studied. A number of studies evaluated the premutation carrier frequency in specific communities. For example, the estimated carrier rate (>55 CGG repeats) in the United States is 1:178,12 whereas in Quebec it was reported to be 1:259–1:397.13 The carrier rate seems to be much lower in the Far East: One study from Japan found no premutations among 947 individuals,14 and the carrier frequency in Taiwan was reported to be as low as 1:1,674.15 By contrast, two studies among Israeli women showed a much higher carrier frequency (1:113 (ref. 16) and 1:157 (ref. 17)) in women with no family history of intellectual disability. There are only a few published studies on differences between carrier rates of expanded alleles (such as premutation alleles) in association with ethnicity. Such differences may be due to a founder effect of repeat length or a founder effect responsible for specific genetic backgrounds/haplotypes characterized by increased or decreased stability upon transmission. For example, a high prevalence of FXS was found among Tunisian Jews from the isle of Djerba, who had a specific unstable allele with no AGG interruptions associated with a rare haplotype.18
The Jewish Israeli population is uniquely composed of multiple ethnic subgroups. The largest group is composed of Jews of Ashkenazi descent. This community was culturally isolated and retained its continuity over a long period of time, leading to the formation of specific founder mutations and increased incidence of certain genetic diseases, such as Tay–Sachs disease and familial dysautonomia.19 Previous studies did not find an increased risk of FXS among Ashkenazi Jews in Israel.18,20 Although no founder effect or increased rate associated with FXS is known among Israeli populations, this disorder has been recommended for genetic screening for females of child-bearing age in Israel because of its high prevalence, and it is the only nonautosomal recessive disorder that is tested as part of the screening tests for common genetic disorders performed in Israel.19
The aim of this study was to evaluate the frequency and characterization of normal, intermediate, and premutation alleles among Ashkenazi versus non-Ashkenazi Jewish women in a large group of Israeli women; to evaluate possible differences in the number of AGG interruptions, in order to assess the risks for FXS and repeat expansion among the Ashkenazi population; and to verify the possible effect of ethnicity on carrier rate and repeat stability.
Materials and Methods
Patients
A total of 15,182 women were tested for FMR1 CGG repeats before or during pregnancy at the Genetic Institute of Tel Aviv Medical Center between 2004 and 2011. These women applied for FMR1 testing on their own initiative or on the advice of their primary-care physician on a self-pay basis. Each woman completed a questionnaire that included information on the ethnic origin of her parents and on her family history of intellectual disability and/or autism spectrum disorders, as well as on FXS. The tests results and the questionnaires were documented in a computerized database from which relevant information for analysis was retrieved. Women with a positive family history for intellectual disability, autism spectrum disorders, or FXS were excluded from the current study. The 11,602 participants were further divided into two groups according to their Jewish ethnic origin, i.e., Ashkenazi or non-Ashkenazi. The Ashkenazi origin was defined based on the reporting of both parents that they were of Ashkenazi origin. Non-Ashkenazi origin was defined based on the reporting of both parents that they were of non-Ashkenazi origin. The women of non-Ashkenazi origin in the study group originated from Morocco, Yemen, Iran, Iraq, the Balkan countries, Tunisia, and/or Libya. Data on origin were given as part of the standard form filled in by each participant. It is important to note that the vast majority of the Jewish population in Israel knows whether their family origin is Ashkenazi or another specific non-Ashkenazi Jewish ethnic group. Women who did not provide information on the origin of both their parents were excluded from the study. Altogether, a total of 2,273 women who had unknown ethnic origin, mixed origin, or whose origins were different from the two specific categories investigated in this study were not included in the ethnicity-based analyses.
CGG repeat analyses
Evaluation of the FMR1 CGG repeat region was performed by various methods, according to the American College of Medical Genetics and Genomics standards and guideline.21 Polymerase chain reaction (PCR)-based analysis was performed in all participants. PCR was performed using an untagged forward primer and a fluorescence-tagged reverse primer, as previously described.22 PCR products were analyzed by GeneScan Analysis software (Applied Biosystems, Carlsbad, CA) according to the manufacturer’s instructions.22 During the period between 2004 and 2009, Southern blot analysis was performed on women with PCR results that demonstrated only a single normal-sized allele or an allele larger than 45 repeats. Briefly, DNA was digested by the restriction enzyme BclI or PstI. A 595-bp PCR-generated probe was used for hybridization.23 Two new PCR-based methods were introduced between 2009 and 2011, and they were used in cases indicated for Southern blot analysis as described earlier. The Abbott Fragile X PCR Kit (Abbott, Abbott Park, IL)24 was used in 2009 and 2010, and the AmplideX FMR1 PCR kit (Asuragen, Austin, TX)25 was used during 2011.
In addition to the CCG repeat length, the PCR products generated with the CGG repeat primer used in the AmplideX kit reveals the number of AGG interspersions in a straightforward manner, as described in detail by Chen et al.25 We therefore used this additional information in the 830 cases that were analyzed by the AmplideX kit and recorded the number of AGG interruptions in addition to the number of CGG repeats in each case.
Alleles were defines as normal (≤44 CGG repeats), intermediate (45–54 CGG repeats), or premutation (45–200 CGG repeats), and women with a premutation allele were defined as FXS carriers.
Statistical analysis
The difference in the frequency of intermediate- and premutation-sized alleles between women of Ashkenazi descent (both parents) and non-Ashkenazi descent (both parents) was assessed using a χ2 test. The difference in the frequency of a reduced number of AGG interruptions in normal- and intermediate-sized alleles was compared between Ashkenazi and non-Ashkenazi women, and the Kolmogorov–Smirnov test was used to determine the difference in the distribution of CGG and AGG repeats between the two groups of women. Significance was set at a P value <0.05.
Results
A total of 11,602 women who had been screened for the FMR1 allelic expansion were included in the study. The vast majority (95.6%) had a normal-sized allele ranging between 7 and 44 CGG repeats. The modal number of CGG repeats in the cases with a normal-sized allele was 30 (50%), with a second peak at 29 (14%) and minor peaks at 20 (6%) and 31 (6%). The prevalence of intermediate-sized alleles (45–54 CGG repeats) was 3.2% (1:31), and the prevalence of premutation-sized alleles (55–200 repeats) was 1.2% (139 women, 1:83). However, most of the premutation-sized alleles (81/139) were within the range of 55–60 repeats. Three women (1:3,867) had the full mutation.
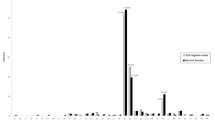
The Ashkenazi group had 4,344 women (both parents of Ashkenazi descent) and the non-Ashkenazi group had 4,985 women (neither parent of Ashkenazi descent). There were differences in the distribution of normal- and intermediate-sized alleles between these two ethnic groups, especially in the minor peaks. Ashkenazi women had a higher rate of 30 repeats and a lower frequency of cases with 33–49 repeats as compared to the non-Ashkenazi women (P < 0.0001; Figure 1 ). A comparison between the rate of intermediate- and premutation-sized alleles revealed that Ashkenazi women had about double the rate of premutation alleles (1:58 vs. 1:104 for the non-Ashkenazi women). However, there was no increase in the frequency of intermediate-sized alleles ( Table 1 ). Notably, there was no increase in the number of alleles in the premutation range with >60 repeats among Ashkenazi women as compared to the non-Ashkenazi women ( Table 1 ).

Distribution of allele sizes <55 repeats among Ashkenazi and non-Ashkenazi women. The proportion of allele distribution by the size of CGG repeats of the FMR1 gene in Ashkenazi and non-Ashkenazi women is described. The x-axis shows the number of CGG repeats, and the y-axis shows the proportion of Ashkenazi versus non-Ashkenazi women for each repeat allele that shows a frequency >1%. White bars: Ashkenazi women, black bars: non-Ashkenazi women.
A total of 830 cases were evaluated for the number of CGG repeats and the number of AGG interruptions during 2011. A total of 760 (91.5%) had a normal-sized allele. There were 326 cases with normal-sized alleles among the Ashkenazi women and 298 cases with normal-sized alleles among the non-Ashkenazi women. The remaining 206 cases were of mixed or unknown origin.
Only 7.5% (25/326) of the Ashkenazi women with normal-sized alleles had only one AGG interruption as compared with 17% (50/298) of the non-Ashkenazi women. Furthermore, whereas only 1.5% (5/326) of the Ashkenazi women who had normal-sized alleles had no interruptions whatsoever, 2.5% (8/298) of the non-Ashkenazi women had no interruptions. Overall, the percentage of women with normal-size alleles and <2 AGG interruptions was 9% in the Ashkenazi women as compared with 19.5% in the non-Ashkenazi women (P = 0.0002, Table 2 ).
Discussion
FXS is considered to be the most common form of inherited intellectual disability, with a variable but high carrier rate. However, the association between the carrier rate and the disease prevalence is not straightforward. Carriers with diverse lengths of CGG repeats have different levels of risk of expansion into a full mutation in female meiosis.8 In addition, interspersed AGG repeats decrease the risk, whereas uninterrupted CGG repeats are associated with allele instability,10 and certain haplotypes are associated with an increased or decreased risk of instability.11
Carrier testing for common genetic disorders is widely used. The tests are selected based on several criteria, including availability, disease frequency, disease severity, age at onset, and lack of effective treatment. These tests are usually associated with common mutations in genes related to autosomal recessive disorders in specific populations. Although FXS has different frequencies in populations in different countries, it is still difficult to evaluate the justification of the performance of population-based tests to identify carriers of the disorder. This study analyzed allele characterization of FMR1 among a large cohort of Ashkenazi and non-Ashkenazi women, in order to better assess the disease risk among the Ashkenazi women and provide tools to assess disease risk in other genetically isolated populations.
The study results demonstrate that the distribution of different repeat alleles is less variable among Ashkenazi women as compared with non-Ashkenazi women, which may be attributed to the increased genetic homogeneity of this population. Similarly, a previous study detected a decreased diversity in the background haplotypes of the FMR1 gene among Ashkenazi Jews, with a higher incidence of a specific DXS548-FRAXAC1-FRAXAC2 haplotype (734+) in the Ashkenazi population.18 Another study25 did not find a founder effect in Ashkenazi patients with FXS but rather detected a high incidence of the 734+ haplotype in the Ashkenazi control group.26 Importantly, this specific haplotype was shown to be associated with a decreased risk of CGG expansion among members of a non-Jewish population.5
The prevalence of premutation alleles in the Israeli population, as detected in this study, is higher than previously reported (1:83 as compared with 1:113 (ref. 16) and 1:157 (ref. 17)). However, in contrast to previous studies, the current study determined ethnicity based on the origin of both parents and included, for statistical purposes, only Jewish women. Comparison between the Ashkenazi and non-Ashkenazi groups revealed that the Ashkenazi women have about a twofold higher prevalence of premutation alleles (1:58 vs. 1:104 for non-Ashkenazi women). This carrier rate was also higher than previously described (1:134) among the Ashkenazi population in the United States.12 Interestingly, the carrier rate among the American Ashkenazi Jews was the highest among all the tested ethnic groups.12
The carrier frequency difference detected between the Ashkenazi and the non-Ashkenazi women was unexpected because there was no difference in the rate of Ashkenazi Jews among FXS patients in Israel.18 There was no significant difference in the rate of alleles with ≥60 repeats between Ashkenazi and non-Ashkenazi women in the current study. Although an allele size of 55–59 CCG repeats is defined as premutation, the risk for expansion to the full mutation in cases with fewer than 60 repeats is <5%,8 and expansion to the full mutation in one generation is rare in cases with fewer than 58 repeats. Indeed, not only is the rate of FXS cases among Ashkenazi Israelis not higher as compared with non-Ashkenazi Israelis,18 it is also likely that these 55–60-repeat alleles are not associated with expansion in the premutation range because the frequency of premutations with >60 repeats among the Ashkenazi and non-Ashkenazi women in our study was similar. Of note, women with a family history positive for intellectual disability, autism spectrum disorders, or FXS were excluded from the study in order to decrease bias. However, because carriers with long repeats have a high risk for affected offspring, these exclusion criteria may lead to an underestimation of the actual carrier rate for the larger repeats in both groups. Evaluation of the number of AGG repeats in the two study groups demonstrated that Ashkenazi women had less than half of the number of cases with <2 AGG interruptions in the normal-sized alleles. Therefore, the repeat alleles would be expected to be more stable among the Ashkenazi women. This suggests that the presence of protective founder alleles with two AGG interruptions is responsible for increased stability of the FMR1 gene among Ashkenazi women.
These results, taken together with the fact that certain haplotypes are protective, suggest that the disease prevalence cannot be estimated solely on the basis of carrier rate, especially in genetically isolated populations. The risk of FXS may be more accurately estimated by disease prevalence than by carrier rates. However, such studies are difficult to perform. Indeed, the common estimation of FXS prevalence at 1:6,000 (ref. 2) is derived from a study that was performed in a select population and is based on the results of screening individuals with intellectual disability rather than the general population. The real prevalence of FXS in various populations and the justification for performing FXS carrier testing for women who are not at a known risk remain uncertain. The current study results show that differences in the prevalence of carriers may be related to ethnicity and that the tendency for instability and expansion may be specific to a population or ethnic group. In addition, the findings suggest that characterizations other than the size of the CGG repeat length, such as ethnicity, haplotypes, and AGG interruptions, may help to better assess risks for offspring with FXS among carriers, especially for those carriers who are at the lower range of CGG repeats.
Disclosure
The authors declare no conflict of interest.
References
Crawford DC, Acuña JM, Sherman SL . FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med 2001;3:359–371.
de Vries BB, van den Ouweland AM, Mohkamsing S, et al. Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Collaborative Fragile X Study Group. Am J Hum Genet 1997;61:660–667.
Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991;65:905–914.
Ashley CT Jr, Wilkinson KD, Reines D, Warren ST . FMR1 protein: conserved RNP family domains and selective RNA binding. Science 1993;262:563–566.
Eichler EE, Holden JJ, Popovich BW, et al. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet 1994;8:88–94.
Fernandez-Carvajal I, Lopez Posadas B, Pan R, Raske C, Hagerman PJ, Tassone F . Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J Mol Diagn 2009;11:306–310.
Zeesman S, Zwaigenbaum L, Whelan DT, Hagerman RJ, Tassone F, Taylor SA . Paternal transmission of fragile X syndrome. Am J Med Genet A 2004;129A:184–189.
Nolin SL, Brown WT, Glicksman A, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet 2003;72:454–464.
Sherman S, Pletcher BA, Driscoll DA . Fragile X syndrome: diagnostic and carrier testing. Genet Med 2005;7:584–587.
Yrigollen CM, Durbin-Johnson B, Gane L, et al. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med 2012;14:729–736.
Curlis Y, Zhang C, Holden JJ, Loesch PK, Mitchell RJ . Haplotype study of intermediate-length alleles at the fragile X (FMR1) gene: ATL1, FMRb, and microsatellite haplotypes differ from those found in common-size FMR1 alleles. Hum Biol 2005;77:137–151.
Hantash FM, Goos DM, Crossley B, et al. FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. Genet Med 2011;13:39–45.
Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K . Prevalence of carriers of premutation-size alleles of the FMRI gene – and implications for the population genetics of the fragile X syndrome. Am J Hum Genet 1995;57:1006–1018.
Otsuka S, Sakamoto Y, Siomi H, et al. Fragile X carrier screening and FMR1 allele distribution in the Japanese population. Brain Dev 2010;32:110–114.
Tzeng CC, Tsai LP, Hwu WL, et al. Prevalence of the FMR1 mutation in Taiwan assessed by large-scale screening of newborn boys and analysis of DXS548-FRAXAC1 haplotype. Am J Med Genet A 2005;133A:37–43.
Toledano-Alhadef H, Basel-Vanagaite L, Magal N, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet 2001;69:351–360.
Berkenstadt M, Ries-Levavi L, Cuckle H, Peleg L, Barkai G . Preconceptional and prenatal screening for fragile X syndrome: experience with 40,000 tests. Prenat Diagn 2007;27:991–994.
Falik-Zakai TC, Shackak E, Yalon M, et al. Predisposition to the fragile X syndrome in Jews of Tunisian descent is due to the absence of AGG interruptions on a rare Mediterranean haplotype. Am J Hum Genet 1997;60:103–112.
Rosner G, Rosner S, Orr-Urtreger A . Genetic testing in Israel: an overview. Annu Rev Genomics Hum Genet 2009;10:175–192.
Dar H, Chemke T, Schaap T, et al. Ethnic distribution of the fragile X syndrome in Israel: evidence of founder chromosomes(?). Isr J Med Sci 1995;31:323–325.
Monaghan KG, Lyon E, Spector EB ; American College of Medical Genetics and Genomics. ACMG Standards and Guidelines for fragile X testing: a revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet Med 2013;15:575–586.
Zhou Y, Lum JM, Yeo GH, Kiing J, Tay SK, Chong SS . Simplified molecular diagnosis of fragile X syndrome by fluorescent methylation-specific PCR and GeneScan analysis. Clin Chem 2006;52:1492–1500.
Rousseau F, Heitz D, Biancalana V, et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med 1991;325:1673–1681.
Juusola JS, Anderson P, Sabato F, Wilkinson DS, Pandya A, Ferreira-Gonzalez A . Performance evaluation of two methods using commercially available reagents for PCR-based detection of FMR1 mutation. J Mol Diagn 2012;14:476–486.
Chen L, Hadd A, Sah S, et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn 2010;12:589–600.
Pesso R, Barkai G, Ravia Y, et al. No founder effect detected in Jewish Ashkenazi patients with fragile-X syndrome. Hum Genet 1997;101:186–189.
Acknowledgements
We thank Esther Eshkol for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Weiss, K., Orr-Urtreger, A., Kaplan Ber, I. et al. Ethnic effect on FMR1 carrier rate and AGG repeat interruptions among Ashkenazi women. Genet Med 16, 940–944 (2014). https://doi.org/10.1038/gim.2014.64
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/gim.2014.64
