
Abstract
Most hybrid zones have existed for hundreds or thousands of years but have generally been observed for only a short time period. Studies extending over periods long enough to track evolutionary changes in the zones or assess the ultimate outcome of hybridization are scarce. Here, we describe the evolution over time of the level of genetic isolation between two karyotypically different species of shrews (Sorex araneus and Sorex antinorii) at a hybrid zone located in the Swiss Alps. We first evaluated hybrid zone movement by contrasting patterns of gene flow and changes in cline parameters (centre and width) using 24 microsatellite loci, between two periods separated by 10 years apart. Additionally, we tested the role of chromosomal rearrangements on gene flow by analysing microsatellite loci located on both rearranged and common chromosomes to both species. We did not detect any movement of the hybrid zone during the period analysed, suggesting that the zone is a typical tension zone. However, the gene flow was significantly lower among the rearranged than the common chromosomes for the second period, whereas the difference was only marginally significant for the first period. This further supports the role of chromosomal rearrangements on gene flow between these taxa.
Similar content being viewed by others

Introduction
Hybrid zones provide ‘natural laboratories’ for investigating the formation (Barton and Hewitt, 1985, 1989; Hewitt, 1988; Harrison, 1993) and maintenance (Jiggins et al., 1997; Rieseberg and Buerkle, 2002) of species differences. Hybrid zones vary greatly in their structure depending on the degree of genetic and ecological differentiation between the two taxa involved, their rates of dispersal and the fitness of hybrid offspring (Harrison, 1993). Moreover, a particularly interesting feature of hybrid zones is that they can move in space and time (Barton and Hewitt, 1981), with significant consequences for both evolutionary and conservation biology. In a recent review, Buggs (2007) stressed the importance of characterizing the spatiotemporal dynamics of hybrid zones. This author emphasized that the movement of a hybrid zone on an ecological timescale can radically alter evolutionary outcomes. Stabilization of the hybrid zone, hybrid speciation, introgression or extinction (through assimilation or outbreeding depression) of one species are outcomes that may result from hybridization and inter-specific recombination (Rieseberg and Wendel, 1993). Hybrid zone stabilization may result from a combination of constant dispersal of parental individuals into the hybrid zone and selection against hybrids (Barton and Hewitt, 1985). Alternatively, Anderson (1948, 1949) argues that a frequent outcome of hybridization is introgression. This process may be uni- or bidirectional and it may be localized or extensive. Species may acquire, through introgression, adaptive characters that allow them to colonize new habitats (Anderson, 1949; Stebbins, 1959; Lewontin and Birch, 1996). It is also possible that if a species has a much higher reproductive success than the other, hybridization may threaten the latter with extinction (Anttila et al., 1998). Observation of hybrid zones over multiple years is the most reliable method for detecting their movement, and evidences of movement have been identified in several genera of both animals and plants (see Buggs, 2007 and references therein).
Recent theoretical studies suggest that chromosomal rearrangements may have a significant role in the limitation of gene flow between species (Noor et al., 2001; Rieseberg, 2001; Navarro and Barton, 2003b), and an increasing amount of data coming from various species seems to show a role of chromosomal changes in speciation. This is the case in studies involving such different lineages as Drosophila, Anopheles, murids or sunflowers (Rieseberg et al., 1995; Noor et al., 2001; Armengol et al., 2005; Ayala and Coluzzi, 2005). An interesting prediction of these models is that chromosomal rearrangements should induce barriers or filters to gene flow that would be specific to some parts of the genome.
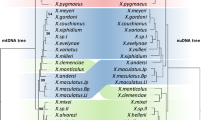
In this study, we investigate the dynamics of a hybrid zone between two shrew species of the Sorex araneus group with different chromosomal rearrangements, and test whether that dynamics vary for different parts of the genome. The closely related shrews of the S. araneus group (Hoffmann, 1971; Meylan and Hausser, 1973; Zima et al., 1998) constitute an illustrative model to test the role of chromosomal rearrangements on reduction of gene flow. Members of this monophyletic group are characterized by karyotypes with low diploid chromosome numbers and a particular sex chromosome system of a XY1Y2/XX type (Sharman, 1956). Autosomal variation between species can be attributed mainly to Robertsonian (Rb) changes accompanied by telomere–centromere tandem translocations, centromere shifts and pericentric inversions (Volobouev and Catzeflis, 1989). The present distributions of the European species of the S. araneus group are largely parapatric and only two species within this group—S. araneus and S. antinorii—are known to hybridize in nature. In this study, we will focus on a hybrid zone located at the Haslital in the Swiss Alps (Innertkirchen, canton Bern). This hybrid zone occurs between Sorex antinorii, which is characterized by the metacentrics gi, hj, kn and l/o (notation: l/o, the slash indicating that the Robertsonian fusion is polymorphic), and the Vaud race of S. araneus, which is characterized by mg, hi, jl, kr and no, according to the nomenclature by Searle et al. (1991) (Figure 1). F1 hybrids between these taxa should be relatively infertile because their chromosomes would assemble into a long chain of 11 elements during meiosis (‘complex’ heterozygotes, CXI; Figure 1) (Narain and Fredga, 1997, 1998; Banaszek et al., 2002). To obtain balanced gametes (with a complete haploid set of chromosome arms) during meiosis, the species-specific chromosomes have to segregate in the same way as when they formed the F1 zygote (Figure 1). As a consequence, only a subset of the produced gametes that comprise a complete haploid set of parental chromosomes will potentially result in viable offspring. Thus, random segregation, recombination and inter-specific gene flow should only occur for the chromosomes, which are similar in both species, whereas species-specific chromosomes should form a linkage block and show relatively little inter-specific gene flow. When comparing the karyotype of the taxa involved in this hybrid zone, it is then possible to define (i) one group of chromosomes similarly arranged as common acrocentrics or metacentrics, and (ii) one group of chromosomes rearranged in different acrocentrics or metacentrics according to the species (Figure 1). Recently, Basset et al. (2006a) used microsatellite loci mapped to individual chromosome arms to test the possibility of an impact of chromosomal rearrangement on gene flow between S. araneus and S. antinorii in the Haslital hybrid zone. These authors showed that inter-specific genetic structure across rearranged chromosomes was indeed larger than that across similarly arranged chromosome (that is, ‘common’ chromosomes). However, the inter-specific difference between the two classes of chromosomes was significant only in the centre of the hybrid zone where the two species lived in sympatry and where there is no evident physical barrier to gene flow. This difference was not significant over a larger geographic range (Basset et al., 2006a) and such results remain difficult to interpret.

Common and rearranged chromosomes in the parent taxa and their combination in F1 hybrids. Black rectangle indicates the complex heterozygote configuration in F1 hybrids for rearranged chromosomes. Asterisks indicate the chromosome arm localization of the markers (see Materials and Methods). Note that the position of locus D24 is indicated in parentheses, as its position is ambiguous (j or l chromosome arm). Arrows stand for segregation ways of the common and rearranged chromosomes during meiosis in F1 hybrid. For further explanations, see text.
At equilibrium, we expect no movement of the zone and no difference over time for the genetic structure estimated over loci located on common and rearranged chromosomes. Alternatively, if such equilibrium is not reached, we expect a higher introgression for loci located on common chromosomes than for those located on rearranged chromosomes. Therefore, we predict a decrease over time of the genetic structure estimated on the common loci, whereas the genetic structure for the rearranged loci must be constant.
In this study, we assess the dynamics of the Haslital hybrid zone over time, by comparing data sampled between 1992 and 1995 (Basset et al., 2006a), with data collected 10 years later (that is, between 2004 and 2005). Our main aim was to detect a hybrid zone movement over time, by studying hybrid zone genetic structure using microsatellite markers, and to compare the genetic structure between common and rearranged chromosomes in the two periods using loci mapped at the chromosome arm level to confirm the pattern observed by Basset et al. (2006a). To our knowledge, this is the first study that addresses the question of differential gene flow in natural populations, by comparing the level of introgression over time on different parts of the genome.
Materials and methods
Sampling
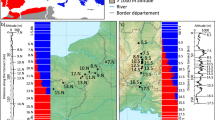
The specimens analysed in this study were sampled during two time periods (Table 1) separated by about 10 years. Sampling was performed along a transect following the Aare river in Haslital Valley (Bern, Central Switzerland, Table 1 and Figure 2). We partitioned the total data set (310 shrews) into two non-overlapping groups, corresponding to individuals sampled in the 1990s (1992–1995) and the 2000s (2004–2005). The capture sessions occurred from September to December of each year, during the period of juvenile dispersal (Churchfield et al., 1995). Shrews were captured alive with Longworth traps filled with hay and baited with larvae of Tenebrio molitor and fresh ground meat. Shrews were toe-clipped before being released on site; tissues were immediately stored in 100% ethanol and kept at −4 °C until DNA extraction.

Study area and sampling localities in the Haslital, Berner Oberland, Switzerland. Pie charts indicate, by locality, the average posterior probability of assignment to a species. White stands for the average posterior probability of assignment to S. araneus, and black stands for the average posterior probability of assignment to S. antinorii.
DNA extraction and microsatellite genotyping
The total genomic DNA was extracted from phalanges using DNeasy Extraction Kit QIAgen (QIAgen, Valencia, CA, USA), resuspended in 200 μl of nanopure water and stored at −20 °C.
Twenty-four microsatellite loci, designated B3, B5, B15 (Basset et al., 2006b), B30, C117, C171, D106, D107, D109, D112, D24 (Basset et al., 2006c) L9, L16, L45, L57, L62, L67, L69 (Wyttenbach et al., 1997), L14, L33, L68, L92 (Balloux et al., 1998) and L13, L99 (Lugon-Moulin et al., 2000) were used. Some individuals have already been genotyped at some loci as part of previous study (Brünner et al., 2002b; Basset et al., 2006a; Yannic et al., 2008a) and were not regenotyped here (see Table 1).
Among the 24 loci, 17 were mapped to the chromosome arm level by Basset et al. (2006c) and the letters in parentheses indicate their chromosome localization: L16 (a), L69 (f), B3 (f), D107 (a), D112 (a), L9 (c), L68 (b), C117 (b), L13 (de), C171 (de), L57 (de), L62 (g), D24 (jl), D106 (h), L99 (n), B30 (o), D109 (o) (see Figure 1). They were used to test for difference in gene flow between common and rearranged chromosomes. The first 11 loci belong to the ‘common group’ and the last six belong to the ‘rearranged group’ (Figure 1).
Polymerase chain reaction conditions are described elsewhere (Wyttenbach et al., 1997; Balloux et al., 1998; Lugon-Moulin et al., 2000; Basset et al., 2006b, 2006c). All PCR were performed in a 20 μl total volume. Cycling was carried out in a PE9700 thermal cycler (Applied Biosystems, Foster City, CA, USA) using the following profile: 95 °C for 5 min, 35 cycles of 30 s at 94 °C, 30 s at annealing temperature (Basset et al., 2006c), 30 s at 72 °C; and a final extension at 72 °C for 4 min. One primer of each pair was labelled with a fluorescent dye (HEX, FAM or NED) on the 5′-end, which allowed analyses on an ABI 377XL sequencer (Applied Biosystems). Data collection, sizing of the bands and analysis were done using the GENESCAN software (Applied Biosystems).
Statistical analyses
Polymorphism, genetic variability of loci and heterozygote deficit within population
The software package FSTAT v. 2.9.4 (updated from Goudet, 1995) was used to calculate allele frequencies, allele numbers, observed heterozygosities (HO) and expected heterozygosities within (HS) and between (HT) samples, following Nei (1987). Heterozygote deficit within populations (FIS>0) was tested using a permutation procedure (10 000 randomizations) to test for non-random mating, using FSTAT.
Genetic structuring and Hierarchical F-statistics
The hierarchical genetic population structure was then investigated using an analysis-of-variance framework. The partition of genetic variance within (FSC) and between (FCT) species was estimated by analysis of molecular variance (Weir and Cockerham, 1984; Michalakis and Excoffier, 1996) using the program ARLEQUIN v. 3.1 (Excoffier et al., 2005), with 10 000 permutations computed for significance values. To assess population differentiation within and between the two species, populations were split by species according to karyotype and/or molecular identification (Brünner et al., 2002b; Basset et al., 2006a, 2007; Yannic et al., 2008a).
Hybrid zone movement and Bayesian assignment
To test for hybrid zone movement, we used a cline analysis following a procedure described by Yannic et al. (2008a). First, we used Bayesian admixture analyses implemented in STRUCTURE 2.2 (Pritchard et al., 2000; Falush et al., 2007), to obtain individual genetic assignment to S. antinorii or S. araneus based on the 24 microsatellite loci and assuming the presence of two populations (K=2). We ran STRUCTURE with 20 repetitions of 100 000 iterations after a burn-in period of 20 000 iterations. Thus, each of the 310 individuals could have been assigned to the S. antinorii cluster (if the posterior probability of assignment q-value was ⩾0.90), to the S. araneus cluster (⩽0.10) or to the inter-specific hybrid group (0.10< q-value<0.90).
Second, we fitted maximum-likelihood clines to Bayesian genetic assignment from 1990s and 2000s data. We kept the estimates of cline shape as simple as possible and only two parameters, cline centre (c) and cline width (w), were estimated following Porter's equations (Porter et al., 1997). These two cline parameters were estimated for the two periods separately. Cline width (w) describes the rate of change in allele frequency in the centre of the zone, where the frequency changes most rapidly, and the centre of a cline (c) is the point where the frequency of alleles switches abruptly and crosses the 0.5 value (Endler, 1977). Therefore, we plotted the change in mean assignment proportion of a population to S. antinorii as given by the Bayesian analyses, along the transect. To test for differences between parameter values, we used the result that doubled the difference in loge likelihood between two models that asymptotically follow a χ2 distribution. Support limits for parameters, such as cline centres and widths, were obtained where the loge likelihood drops to two units below the maximum. These support limits (SL) are asymptotically equivalent to 95% confidence interval.
Genetic structuring for common and rearranged chromosomes
The partition of genetic variance within (FSC) and between (FCT) species for common and rearranged chromosomes was estimated across all populations sampled in the 2000s by an analysis of molecular variance implemented in the program ARLEQUIN v. 3.1 (Excoffier et al., 2005), with 10 000 permutations for significance values. Results were then compared with those obtained by Basset et al. (2006a) for the 1990 period and reanalyzed here. Intra-specific structure (FSC) was used as a control for the real significance of observed differences between species because there are no karyotypic differences within species. Differences of genetic structure obtained on the common and rearranged chromosomes within a sampling period were tested by permutation tests consisting of 10 000 permutations between the two groups of chromosomes (11 loci on common chromosomes and 6 loci on rearranged chromosomes).
Results
Polymorphism, genetic variability of loci and heterozygote deficit within population
The number of total alleles and species-specific alleles as well as the observed and expected heterozygosities from the 1990s and 2000s hybrid zones are detailed in Table 2 and Supplementary Tables (available online). We found a larger proportion of species-specific alleles in the 1990s (57.0%) than in the 2000s (51.4%) (nas1990s=243/426 vs nas2000s=266/517; Fisher's exact test, P<0.05). This difference is mainly caused by the higher S. antinorii species-specific alleles in the 1990s (46.7%) than in the 2000s (39.5%; Fisher's exact test, P<0.05), whereas this proportion was stable for S. araneus (29.6 and 30.6%, respectively; Fisher's exact test, P=0.48).
For the 1990s, the number of alleles per locus ranged from 3 to 34 (Table 2). Expected heterozygosities within samples (HS) ranged from 0.37 to 0.91, with an average of 0.82, whereas expected heterozygosities between samples (HT) averaged 0.82 (0.54–0.95; Table 2). Observed heterozygosities (HO) ranged from 0.22 to 0.93. The average value was 0.65 (Table 2).
For the 2000s, the number of alleles per locus ranged from 5 to 46 (Table 2). Expected heterozygosities within samples (HS) ranged from 0.34 to 0.93, with an average of 0.78, whereas expected heterozygosities between samples (HT) averaged 0.83 (0.59–0.95; Table 2). Observed heterozygosities (HO) ranged from 0.13 to 0.93. The average value was 0.63 (Table 2).
Across all loci, within-population heterozygote deficit was highly significantly different from 0 for the two time periods (FIS-1990s=0.171, P<0.001; and FIS-2000s=0.197, P<0.001; Table 2). A part of this deficit could be explained by the presence of genotyping errors or null alleles (Van Oosterhout et al., 2004). Therefore, we used MICROCHECKER version 2.2.3 (Van Oosterhout et al., 2004) to test whether potential genotyping errors or null alleles could affect our results. As our results were qualitatively similar when using either the raw genotypes or adjusted genotypes according to MICROCHECKER (results not shown), we present only the results based on observed frequencies.
Genetic structuring and hierarchical F-statistics
The overall high fixation indexes suggested a moderate but significant and stable genetic structure over time (FST-1990s=0.177, P<0.001; and FST-2000s=0.167, P<0.001). Whereas the most important part of the variance was found between species (FCT-1990s=0.152, P<0.001 and FCT-2000s=0.139, P<0.001), the variance among populations within species was much lower but still significant (FSC-1990s=0.030, P<0.001; and FSC-2000s=0.032, P<0.001). Results were in the same order of magnitude over time (Table 3).
Hybrid zone movement and Bayesian assignment
Bayesian admixture analyses of autosomal microsatellites assigned 58% (67/115) for the 1990s and 57% (111/195) for the 2000s of the individuals to S. antinorii at a probability >0.90. A total of 31% of the shrews (36/115) were assigned to S. araneus for the 1990s, and 38% (74/195) for the 2000s. The remaining shrews, 10.4% (12/115) for the 1990s and 5.1% (10/195), were inter-specific hybrids (0.103⩽qind⩽0.855 for the 1990s and 0.107⩽qind⩽0.793 for the 2000s). Over time, there is no evidence of change in average posterior probability of assignment to a species (Figure 2). The proportion of each species in each sampling location does not differ significantly between the 1990s and the 2000s (rank Wilcoxon's signed rank test, W=26, P=0.90) and are highly correlated (R2=0.99, P<0.001).
The analysis of cline shape for the combined 24 markers showed no evidence for a hybrid zone movement. Cline shape parameter calculated for the two periods and the relationship between Bayesian genetic assignment and geographic position in the hybrid zone transect are shown in Figure 3. The cline centre information is of particular importance, as it shows the position of the hybrid zone over the course of about 10 years. Cline centres and widths obtained by fitting maximum-likelihood curves did not differ significantly between the two periods and were overall coincident (that is, same centres) and concordant (that is, same width). We obtained cline centre estimates for the 1990s, c=6.51 km (95% CI 6.12–6.81), and for the 2000s, c=6.25 km (95% CI 5.81–6.65). The cline width estimates were for the 1990s, w=2.42 km (95% CI 1.77–3.90), and for the 2000s, w=3.21 km (95% CI 2.23–4.75). Two populations were analysed in the 2000s, but not in the 1990s (that is, localities 2 and 9), but excluding these localities from the analyses did not affect our results. Finally, all likelihood ratio tests for difference in cline shape were not significant, suggesting that the hybrid zone remained fairly stable during the period analysed.

Scatter plots of S. antinorii allele frequencies versus geographical position (km) for the two periods in the Haslital hybrid zones hybrid zone between S. antinorii and S. araneus. Dots are observed frequencies and lines are fitted curves.
Genetic structuring for common and rearranged chromosomes
The genetic structure estimated across the common and rearranged loci was highly significant for the two categories of loci and for the two periods (see Table 4). The FST values estimated across rearranged loci are about three times higher than values obtained across common loci in both time periods, and the differences are significant (permutation tests: P<0.05 for the 1990s and 2000s). Hierarchical analyses showed that for both periods, the genetic differentiation within each species (FSC) is in the same order of magnitude for common and rearranged chromosome and do not differ significantly (permutation tests: P>0.05 for both the 1990s and 2000s). However, the genetic differentiation between species (FCT) was highly significant across common and rearranged loci (see Table 4). Moreover, for the 1990s, this genetic differentiation was three times higher across rearranged than across common loci (FCT-Common=0.082 vs FCT-Rearranged=0.248), but the difference between these two groups was not significant (permutation test: P<0.10). In contrast, for the 2000s, this genetic differentiation was more than four times higher across rearranged than across common loci (FCT-Common=0.067 vs FCT-Rearranged=0.290), and the difference between these two groups was significant (permutation test: P<0.05). This result clearly supports the hypothesis that in this hybrid zone, chromosomal rearrangements act as a barrier to gene flow for only some parts of the genome. Permutation tests performed on the different fixations indices did not show differences in genetic structure over time (permutation tests: FST-1990s vs FST-2000s, P=0.87; FSC-1990s vs FSC-2000s, P=0.70; and FCT-1990s vs FCT-2000s, P=0.69).
Discussion
The outcome of hybridization between two genetically differentiated species is difficult to predict. There is a variety of outcomes that may result from hybridization, including the reintegration of the two previously distinct gene pools, the formation of stable hybrid zone or the completion of speciation by reinforcement (Rieseberg and Wendel, 1993). This depends fundamentally on the fitness of hybrids and the life-history characteristics of the hybridizing species (Carney et al., 2000). We found that the hybrid zone between S. araneus and S. antinorii is geographically stable over time. This suggests that this zone is a typical tension zone where its stabilization may result from a combination of dispersal of individuals from the parental populations into the hybrid zone and selection against hybrids (Barton and Hewitt, 1985; Hewitt, 1988). Moreover, our data support the hypothesis of chromosomal rearrangements playing a role in the reduction of gene flow between S. antinorii and S. araneus, confirming the pattern observed by Basset et al. (2006a) in the 1990s’ period.
Overall genetic structure over time
Using 24 microsatellite markers, we found similar overall genetic structure in the S. araneus—S. antinorii hybrid zone over time. Similarly, cline shape analyses supported stability, as the cline parameters between the two periods were coincident (that is, same centre) and concordant (that is, same width). Cline width provides the most direct information about reproductive isolation because it measures the extent of gene flow on the centre of the zone. Nevertheless, we found a larger proportion of species-specific alleles in the 1990s (57.0%) than in the 2000s (51.4%). This difference is mainly caused by the higher S. antinorii species-specific alleles in the 1990s (46.7%) than in the 2000s (39.5%), whereas this proportion was stable for S. araneus (29.6 and 30.6%, respectively, see Results). Such a pattern may be evidence for higher inter-specific gene flow in the 2000s than in the 1990s or by a recent advance of S. antinorri into the S. araneus territory as suggested by the overall higher proportion of S. antinorii specific alleles. The localization of a tension zone is affected by a variety of forces. In particular, these zones tend to move to local regions of low hybrid fitness, low dispersal or low population density (Barton and Hewitt, 1985). In addition, if there were some behavioural, ecological or other superiority of one of the hybridizing taxa, the zone would move in favour of the superior taxon (Barton and Hewitt, 1981, 1985). The absence of introgression or apparent movement of the Haslital zone over a 10-year period does not necessarily mean that the hybrid zone stayed at the same place because of secondary contact between the two species. The hybrid zone is at present situated in the lower part of the valley. However, it is most likely that S. antinorii progressively moved down from the Grimsel Pass (2165 m) into the S. araneus territory, as this pass is the only possible southern recolonization way after the retreat of the glaciers (see Figure 2). A radiocarbon date of the earliest organic sediment deposition on the Grimsel Pass shows that this high elevation pass was ice-free by approximately 9000–12 000 years BP (Kelly et al., 2002). Therefore, S. antinorii and S. araneus might have already been in contact in the Haslital for several hundred or thousand years. Thus, the 10 years between the two sampling periods might be too small on an evolutionary time scale to accurately detect hybrid zone movement.
Several indirect indications may indicate that a hybrid zone is still moving. For example, in a sister hybrid zone between S. araneus race Cordon and S. antinorii located in the French Alps, the pattern of introgression of the mtDNA of S. araneus race Cordon into the genetic background of S. antinorii suggests hybrid zone movement (Balloux et al., 2000; Yannic et al., 2008a). Although this asymmetrical introgression could be due to selective incorporation of S. araneus haplotype at the hybrid zone, the introgressed haplotypes could also be the result of an eastward hybrid zone movement in favour of S. antinorii. This hypothesis is in agreement with the probable routes used by S. antinorii to recolonize the northern part of its home range from southern Italy after the last maximum glaciations (Yannic et al., 2008c). However, short-term studies over a 4-year period of this hybrid zone have not shown ongoing movement (Lugon-Moulin et al., 2000).
An important difference between the Haslital and Les Houches hybrids zones is the geographical location of the zone. At Les Houches, the contact zone is located at a mountain river (Brünner and Hausser, 1996). It was assumed that cline variation and genetic differentiation between the two species in the centre of this zone is the synergetic product of genetic incompatibilities and of the dispersal-reducing effect of the river (Lugon-Moulin et al., 1999a, 1999b; Brünner et al., 2002b). Thus, genetic differentiation would most likely be smaller if the river was not preventing the co-occurrence of two species. In Haslital, both the species apparently live in sympatry and there is no evident geographical barrier (for example, rivers) between species, although subtle ecological differences are difficult to detect. In consequence, the relative stability of the Haslital hybrid zone suggests that a strong genetic barrier to gene flow between the two species governs this zone. This was further supported by the very narrow clines observed in this zone for chromosomes (Brünner et al., 2002b), mitochondrial and Y-chromosome-linked markers (Yannic et al., 2008a).
Differences over time between common and rearranged chromosomes
Among the different factors limiting gene flow between species, it is often proposed that chromosomal rearrangements have an impact on hybrid fitness. Here, S. araneus and S. antinorii differ in their karyotypic composition because of differences in Robertsonian fusions (for a review on consequences of chromosomal incompatibilities in S. araneus, refer the review by Searle and Wójcik, 1998). Recent studies showed that chromosomal rearrangements probably limit the gene flow between S. araneus and S. antinorii (Brünner et al., 2002b; Basset et al., 2006a). By studying the 17 microsatellite markers located on common and rearranged chromosomes (11 located on common chromosomes and 6 located on rearranged chromosomes), we tested the role of chromosomal rearrangements on the genetic structure of this zone. We found a significantly higher level of genetic structure across loci located on the rearranged chromosomes than across loci located on common chromosomes. This finding strengthened the results obtained by Basset et al. (2006a) for the 1990s sampling period, as we found a significantly higher inter-specific structure for the rearranged chromosomes on the whole hybrid zone. In the 1990s, this difference was significant only in the centre of hybrid zone where the impact of chromosomal rearrangements on gene exchange is thought to be the stronger. Differences between the two studies may result from a higher statistical power due to sampling a relatively higher number of localities (7 localities for the 1990s vs 9 localities for the 2000s) and higher numbers of individuals per locality (16.4 shrews for the 1990s vs 22.8 shrews for the 2000s) or reinforcement of differentiation between the two species. The intra-specific structure, which can be used as a control of the role of rearrangements as there is no karyotypic difference within species, did not show a significant difference between the common and rearranged chromosomes within species in both time periods. After secondary contact between S. antinorii and S. araneus, gene flow probably reduced inter-specific differences for most regions of the genome. However, the large linkage blocks formed by rearranged chromosomes in karyotypic hybrids most likely prevent the introgression of heterospecific alleles between species. This reduction of gene flow for the rearranged part of the genome may be explained by the one-way segregation of rearranged chromosomes as well as by the reduction of recombination among these same chromosomes.
Implication for speciation
This study provides an important confirmation for the theoretical prediction that chromosomal rearrangements reduce gene flow in inter-specific hybrid zones. Chromosomal rearrangements, acting as a barrier to gene flow, prevent fusion of populations coming into contact, by causing a reduction in hybrid viability or fertility (the ‘hybrid dysfunction’ models, Ayala and Coluzzi, 2005) or a reduction in recombination among the rearranged chromosomes (the ‘suppressed recombination’ models, Ayala and Coluzzi, 2005). In this study, it is not possible to conclude on the respective role of such mechanisms in reducing gene flow. Data from the S. araneus group suggest that Robertsonian heterozygotes do not suffer from infertility as substantially as other taxa (Searle, 1993; Narain and Fredga, 1997, 1998; Banaszek et al., 2000). In addition, several studies of hybrid zones between karyotypic races of S. araneus have shown a low level of genetic differentiation among races (for example, Abisko/Sidensjö, FST=0.015, Andersson et al., 2004; Drnholec/Bialowieza, FST=0.021, Jadwiszczak et al., 2006; or Uppsala/Hällefors, FST=0.026, Wyttenbach et al., 1999). Thus, chromosomal rearrangements do not seem to impede gene flow in these zones, although data on specific gene flow across loci located on common and rearranged chromosomes are missing. The situation is quite different in the inter-specific hybrid zone addressed in this study. The two species had a long period of independent evolution (Searle and Wójcik, 1998; Brünner et al., 2002a) and probably diverged karyotypically and genetically in distinct refugia: in the Apennine peninsula for S. antinorii and most probably in the Balkans or the region between Carpathians and the Black Sea for S. araneus (Taberlet et al., 1994; Brünner et al., 2002a; Yannic et al., 2008b). Therefore, it is most likely that both genetic incompatibilities accumulated in allopatry and chromosomal rearrangements act as a reproductive barrier (Rieseberg et al., 1999; Noor et al., 2001; Navarro and Barton, 2003a; Basset et al., 2006a). Nevertheless, rearranged chromosomes form a large linkage block that may carry and link genes important for reproductive isolation. Therefore, even if there is little evidence that chromosomal rearrangements cause speciation per se, they probably help in maintaining reproductive isolation and thus independent evolution of the two species.
General conclusion
The role of chromosome in speciation is still much debated. Lower gene exchanges or higher genetic divergences across genomic regions differing by chromosomal rearrangements have been detected between species of flies (Noor et al., 2001; Machado et al., 2002; Ortiz-Barrientos et al., 2002) or sunflowers (Rieseberg et al., 1999), between chromosomal races of house mouse (Panithanarak et al., 2004) or between human and chimpanzee (Navarro and Barton, 2003b). However, species for which genetic maps are available (for example, sunflower, house mice or humans) are scarce, and additional empiric studies from non-model species are of primary importance. In such a context, our study add to other studies to show the utility of mapped loci for such analyses and provide evidence of reduced gene flow at some specific parts of the genome (that is, between rearranged chromosomes).
References
Anderson E (1948). Hybridization of the habitat. Evolution 2: 1–9.
Anderson E (1949). Introgressive Hybridization. John Wiley and Sons: New York.
Andersson AC, Narain Y, Tegelstrom H, Fredga K (2004). No apparent reduction of gene flow in a hybrid zone between the West and North European karyotypic groups of the common shrew, Sorex araneus. Mol Ecol 13: 1205–1215.
Anttila C, Daehler C, Rank N, Strong D (1998). Greater male fitness of a rare invader (Spartina alterniflora, Poaceae) threatens a common native (Spartina foliosa) with hybridization. Am J Bot 85: 1597–1601.
Armengol L, Marques-Bonet T, Cheung J, Khaja R, Gonzalez J, Scherer S et al. (2005). Murine segmental duplications are hot spots for chromosome and gene evolution. Genomics 86: 692–700.
Ayala FJ, Coluzzi M (2005). Chromosome speciation: humans, drosophila, and mosquitoes. Proc Natl Acad Sci USA 102: 6535–6542.
Balloux F, Ecoffey E, Fumagalli L, Goudet J, Wyttenbach A, Hausser J (1998). Microsatellite conservation, polymorphism, and GC content in shrews of the genus Sorex (Insectivora, Mammalia). Mol Biol Evol 15: 473–475.
Balloux F, Lugon-Moulin N, Hausser J (2000). Estimating gene flow across hybrid zones: how reliable are microsatellites? Acta Theriologica 45: 93–101.
Banaszek A, Fedyk S, Fiedorczuk U, Szalaj KA, Chetnicki W (2002). Meiotic studies of male common shrews (Sorex araneus L.) from a hybrid zone between chromosome races. Cytogenet Genome Res 96: 40–44.
Banaszek A, Fedyk S, Szalaj KA, Chetnicki W (2000). A comparison of spermatogenesis in homozygotes, simple Robertsonian heterozygotes and complex heterozygotes of the common shrew (Sorex araneus L.). Heredity 85: 570–577.
Barton NH, Hewitt GM (1981). The genetic-basis of hybrid inviability in the grasshopper Podisma pedestris. Heredity 47: 367–383.
Barton NH, Hewitt GM (1985). Analysis of hybrid zones. Annu Rev Ecol Systemat 16: 113–148.
Barton NH, Hewitt GM (1989). Adaptation, speciation and hybrid zones. Nature 341: 497–503.
Basset P, Yannic G, Brünner H, Hausser J (2006a). Restricted gene flow at specific parts of the shrew genome in chromosomal hybrid zones. Evolution 60: 1718–1730.
Basset P, Yannic G, Hausser J (2006b). Genetic and karyotypic structure in the shrews of the Sorex araneus group: are they independent? Mol Ecol 15: 1577–1587.
Basset P, Yannic G, Hausser J (2007). Using a Bayesian method to assign individuals to karyotypic taxa in shrew hybrid zones. Cytogenet Genome Res 116: 282–288.
Basset P, Yannic G, Yang FT, O’Brien PCM, Graphodatsky AS, Ferguson-Smith MA et al. (2006c). Chromosome localization of microsatellite markers in the shrews of the Sorex araneus group. Chromosome Res 14: 253–262.
Brünner H, Hausser J (1996). Genetic and karyotypic structure of a hybrid zone between the chromosomal races Cordon and Valais in the common shrew, Sorex araneus. Hereditas 125: 147–158.
Brünner H, Lugon-Moulin N, Balloux F, Fumagalli L, Hausser J (2002a). A taxonomical re-evaluation of the Valais chromosome race of the common shrew Sorex araneus (Insectivora: Soricidae). Acta Theriologica 47: 245–275.
Brünner H, Lugon-Moulin N, Hausser J (2002b). Alps, genes, and chromosomes: their role in the formation of species in the Sorex araneus group (Mammalia, Insectivora), as inferred from two hybrid zones. Cytogenet Genome Res 96: 85–96.
Buggs RJA (2007). Empirical study of hybrid zone movement. Heredity 99: 301–312.
Carney SE, Gardner KA, Rieseberg L (2000). Evolutionary changes over the fifty year history of a hybrid population of sunflowers (Helianthus). Evolution 54: 462–474.
Churchfield S, Hollier J, Brown K (1995). Population dynamics and survivorship in the common shrew Sorex araneus in southern England. Acta Theriologica 40: 53–68.
Endler JA (1977). Geographic Variation, Speciation, and Clines. Princeton University Press: Princeton, New Jersey.
Excoffier L, Laval G, Schneider S (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1: 47–50.
Falush D, Stephens M, Pritchard JK (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578.
Goudet J (1995). FSTAT (Version 1.2): a computer program to calculate F-statistics. J Heredity 86: 485–486.
Harrison R (1993). Hybrid Zones and the Evolutionary Process. Oxford University Press: Oxford.
Hewitt GM (1988). Hybrid zones—natural laboratories for evolutionary studies. Trends Ecol Evolut 3: 158–167.
Hoffmann R (1971). Relationships of certain Holartic shrews, genus Sorex. Zeitschrift für Säugetierkunde 36: 193–200.
Jadwiszczak KA, Ratkiewicz M, Banaszek A (2006). Analysis of molecular differentiation in a hybrid zone between chromosomally distinct races of the common shrew Sorex araneus (Insectivora : Soricidae) suggests their common ancestry. Biol J Linnean Soc 89: 79–90.
Jiggins C, McMillan W, King P, Mallet J (1997). The maintenance of species differences across a Heliconius hybrid zone. Heredity 79: 495–500.
Kelly M, Schlüchter C, Kubik PW (2002). Surface exposure dating of glacially eroded bedrock on the Grimsel pass, Swiss alps. In: Annual Report. Physics EPIB (ed). EPF Zurich: Zurich, p. 1.
Lewontin R, Birch L (1996). Hybridization as a source of variation for adaptation to a new environment. Evolution 20: 315–336.
Lugon-Moulin N, Balloux F, Hausser J (2000). Genetic differentiation of common shrew Sorex araneus populations among different alpine valleys revealed by microsatellites. Acta Theriologica 45: 103–117.
Lugon-Moulin N, Brünner H, Balloux F, Hausser J, Goudet J (1999a). Do riverine barriers, history or introgression shape the genetic structuring of a common shrew (Sorex araneus) population? Heredity 83: 155–161.
Lugon-Moulin N, Brünner H, Wyttenbach A, Hausser J, Goudet J (1999b). Hierarchical analyses of genetic differentiation in a hybrid zone of Sorex araneus (Insectivora: Soricidae). Mole Ecol 8: 419–431.
Machado CA, Kliman RM, Markert JA, Hey J (2002). Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Mol Biol Evolut 19: 472–488.
Meylan A, Hausser J (1973). Les chromosomes des Sorex du groupe araneus-arcticus (Mammalia, Insectivora). Zeitschrift für Säugetierkunde 38: 143–158.
Michalakis Y, Excoffier L (1996). A genetic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142: 1061–1064.
Narain Y, Fredga K (1997). Meiosis and fertility in common shrews, Sorex araneus, from a chromosomal hybrid zone in central Sweden. Cytogenet Cell Genet 78: 253–259.
Narain Y, Fredga K (1998). Spermatogenesis in common shrews, Sorex araneus, from a hybrid zone with extensive Robertsonian polymorphism. Cytogenet Cell Genet 80: 158–164.
Navarro A, Barton N (2003a). Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution 57: 447–459.
Navarro A, Barton NH (2003b). Chromosomal speciation and molecular divergence—accelerated evolution in rearranged chromosomes. Science 300: 321–324.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Noor MAF, Grams KL, Bertucci LA, Reiland J (2001). Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci USA 98: 12084–12088.
Ortiz-Barrientos D, Reiland J, Hey J, Noor MAF (2002). Recombination and the divergence of hybridizing species. Genetica 116: 167–178.
Panithanarak T, Hauffe HC, Dallas JF, Glover A, Ward RG, Searle JB (2004). Linkage-dependent gene flow in a house mouse chromosomal hybrid zone. Evolution 58: 184–192.
Porter AH, Wenger R, Geiger H, Scholl A, Shapiro AM (1997). The Pontia daplidice-edusa hybrid zone in northwestern Italy. Evolution 51: 1561–1573.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rieseberg L, Buerkle C (2002). Genetic mapping in hybrid zones. Amer Naturalist 159: S36–S50.
Rieseberg L, Vanfossen C, Desrochers A (1995). Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature 375: 313–316.
Rieseberg LH (2001). Chromosomal rearrangements and speciation. Trends Ecol Evolut 16: 351–358.
Rieseberg LH, Wendel J (1993). Introgression and its consequences in plants. In: Harrison RG (ed). Hybrid Zones and the Evolutionary Process. Oxford University Press: New York, pp 70–109.
Rieseberg LH, Whitton J, Gardner K (1999). Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152: 713–727.
Searle J (1993). Chromosomal hybrid zones in eutherian mammals. In: Harrison RG (ed). Hybrid Zones and the Evolutionary Process. Oxford University Press: New York, pp 309–353.
Searle J, Wójcik J (1998). Chromosomal evolution: the case of Sorex araneus. In: Wójcik J, Wolsan M (eds). Evolution of Shrews. Mammal Research Institute, Polish Academy of Sciences: Bialowieza, pp 219–268.
Searle JB, Fedyk S, Fredga K, Hausser J, Volobouev VT (1991). Nomenclature for the chromosomes of the common shrew (Sorex araneus). Mémoires de la Société Vaudoise de Sciences Naturelles 19: 13–22.
Sharman GB (1956). Chromosomes of the common shrew. Nature 177: 941–942.
Stebbins GLJ (1959). The role of hybridization in evolution. Proc Amer Philos Soc 103: 213–251.
Taberlet P, Fumagalli L, Hausser J (1994). Chromosomal versus mitochondrial-DNA evolution—tracking the evolutionary history of the Southwestern European populations of the Sorex araneus group (Mammalia, Insectivora). Evolution 48: 623–636.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004). MicroCheckers: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Volobouev V, Catzeflis F (1989). Mechanisms of chromosomal evolution in 3 European species of the Sorex araneus-arcticus group (Insectivora, Soricidae). Zeitschrift Fur Zoologische Systematik und Evolutionsforschung 27: 252–262.
Weir B, Cockerham C (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wyttenbach A, Favre L, Hausser J (1997). Isolation and characterization of simple sequence repeats in the genome of the common shrew. Mol Ecol 6: 797–800.
Wyttenbach A, Goudet J, Cornuet JM, Hausser J (1999). Microsatellite variation reveals low genetic subdivision in a chromosome race of Sorex araneus (Mammalia, Insectivora). J Heredity 90: 323–327.
Yannic G, Basset P, Hausser J (2008a). A hybrid zone with coincident clines for autosomal and sex-specific markers in the Sorex araneus group. J Evol Biol 21: 658–667.
Yannic G, Basset P, Hausser J (2008b). A new perspective on the evolutionary history of western European Sorex araneus group revealed by paternal and maternal molecular markers. Mol Phylogenet Evol 47: 237–250.
Yannic G, Basset P, Hausser J (2008c). Phylogeography and recolonization of the Swiss Alps by the Valais shrew (Sorex antinorii), inferred with autosomal and sex-specific markers. Mol Ecol 17: 4103–4118.
Zima J, Lukáčová L, Macholán M (1998). Chromosomal evolution in shrews. In: Wójcik J, Wolsan M (eds). Evolution of Shrews. Mammal Research Institute, Polish Academy of Sciences: Bialowieza, pp 175–218.
Acknowledgements
We thank Véronique Helfer and Emmanuelle Yannic for help in the field; Lucie Büchi, Tanja Schwander, Valerie Vogel and anonymous referees for insightful comments on the manuscript; and Allan McDevitt for help with English. This work was supported by Fondation Agassiz (Switzerland) and Société Académique Vaudoise (Switzerland) grants to GY.
Author information
Authors and Affiliations
Corresponding author
Additional information
Licence statement
This study complied with the current laws of Switzerland.
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Yannic, G., Basset, P. & Hausser, J. Chromosomal rearrangements and gene flow over time in an inter-specific hybrid zone of the Sorex araneus group. Heredity 102, 616–625 (2009). https://doi.org/10.1038/hdy.2009.19
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/hdy.2009.19
Keywords
This article is cited by
-
Unraveling the effect of genomic structural changes in the rhesus macaque - implications for the adaptive role of inversions
BMC Genomics (2014)
-
Chromosome rearrangements, recombination suppression, and limited segregation distortion in hybrids between Yellowstone cutthroat trout (Oncorhynchus clarkii bouvieri) and rainbow trout (O. mykiss)
BMC Genomics (2013)
-
Spatio-temporal variation in the structure of a chromosomal polymorphism zone in the house mouse
Heredity (2012)
-
Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species
Heredity (2009)
