
Abstract
We aimed to reveal the effects of range expansion and subsequent lineage admixture from separated glacial refugia on genetic diversity of Kalopanax septemlobus in Japan, by combining nuclear microsatellite data and ecological niche modelling. Allelic richness and gene diversity were compared at the population and regional level. We also statistically examined these indices as a function of population accessibility to the last glacial maximum (LGM) palaeodistribution reconstructed by ecological niche modelling to test a simple range expansion scenario from glacial refugia. Genetic diversity was highest in the populations of southern Japan and gradually decreased towards the north. However, an additional centre of genetic diversity, when measured as gene diversity, was found in northern Honshu Island, where distinct lineages were shown to be in contact. Positive effects of population accessibility to the LGM range were detected in both diversity indices at different spatial scales. The combined data support independent postglacial range expansions towards the north from the edge populations on the exposed coastal shelf of Pacific and Sea of Japan in northern Honshu during the LGM, which subsequently resulted in markedly low genetic diversity in the northernmost extant range, Hokkaido. The regional increase in gene diversity in northern Honshu is likely to be the result of postglacial lineage admixture. Relative difference in the spatial scales best relating population genetic diversity with the LGM distribution can be explained by a higher rate of allelic richness diversity loss during range expansions and stronger effects of lineage admixture on gene diversity.
Similar content being viewed by others

Introduction
During the late Quaternary (0.9 Myr ago–present), the amplified 0.1 Myr glacial–interglacial climate cycles was omnipresent on a global scale (Petit et al., 1999; Lisiecki and Raymo, 2005). These climatic oscillations produced great changes in species distribution; that is, some species responded by dispersing to new locations, whereas others survived in refugia during unfavourable periods and subsequently were able to re-expand their range (Hewitt, 1996, 2000). These distributional changes have left ‘footprints’ in the genetic structure of many temperate tree species by impacting the geographic distributions of genetic diversity and intraspecific lineages (Tomaru et al., 1997; Petit et al., 2003b; Heuertz et al., 2004; McLachlan et al., 2005; Magri et al., 2006; Gao et al., 2007; Tsumura et al., 2007). In the northern hemisphere, many temperate tree species are thought to have mostly been exterminated during the last glacial in the northern parts of their current ranges, whereas populations in the southern parts are thought to have been able to persist through multiple climate cycles. The current northern populations, therefore, are likely to display the genetic structure strongly influenced by postglacial colonisation during the Holocene. Two factors are likely to be more important in having shaped the genetic structure of these populations; founding events and the admixture of genetic lineages from separate source populations. Founding events most often result in the loss of genetic diversity and increases in genetic differentiation from source populations (Le Corre and Kremer, 1998) with rare long-distance dispersal events accelerating these founder effects compared with simple diffuse dispersal scenarios (Austerlitz and Garnier-Gere, 2003). On the other hand, the admixture of intraspecific lineages during postglacial colonisation may lessen the founder effect or possibly even increase genetic diversity in comparison with the diversity of source populations (Comps et al., 2001; Walter and Epperson, 2001; Heuertz et al., 2004).
Phylogeography combined with independent evidences is a powerful approach to improve our understanding of the past distributional changes of plant species. For example, fossil records combined with genetic data allowed detailed inferences to be made about the pattern of postglacial spread from multiple glacial refugia in Fagus sylvatica (Magri et al., 2006). Even for a species with scant fossil records, palaeovegetation reconstructions based on the fossil record may be helpful for locating glacial refugia, provided that the species coexisted with the same species during glacial periods as they do at present. However, because each species is thought to have unique ecological niches limited by different environmental factors, the combination of climatic variables that existed during the last glacial maximum (LGM, ca. 21 000yr BP) may have led to community structures that differed from those in existence today (Carstens and Richards, 2007). Therefore, it cannot be assumed that all species that co-occur at present were similarly affected by past climatic changes. Ecological niche models can also provide a source of evidence independent from genetic data, which is particularly pertinent for species that are poorly represented in the fossil records. Once modern distribution data and environmental variables are collated in an ecological niche model, historical species distributions can be approximated by projecting species parameters onto a simulated past climate (Svenning et al., 2008). Thus, when these two approaches are combined, understandings of the past processes having shaped population genetic diversity and structure can be greatly improved (Peterson et al., 2004; Carstens and Richards, 2007; Waltari et al., 2007; Solomon et al., 2008).
The Japanese Archipelago comprises four mountainous main islands 30–45°N (from north to south: Hokkaido, Honshu, Shikoku and Kyushu) and thousands of small islands, including the Ryukyu Archipelago to 24°N. It has numerous geographical barriers (for example, high mountain ranges, valleys and seaways), which are expected to have influenced the distribution patterns and past migration of many organisms in Japan, including tree species. According to pollen and macrofossil records, temperate deciduous forest is thought to have retreated to southern coastal refugial areas of the archipelago (<38°N) during the LGM (Tsukada, 1985). At the end of the last glacial, temperate deciduous forest biome is thought to have began to expand its range towards northern areas, having replaced the boreal coniferous forests and tundra vegetation that existed in these locations during the LGM (Tsukada, 1983).
Kalopanax septemlobus (Thunb.) Koidz. (Araliaceae) (Ohashi, 1994; Ohba, 1999; Xiang and Lowry, 2007), castor aralia, is a deciduous broadleaf tree species, occurring mainly in the temperate deciduous forests throughout Japan from 24°N in Okinawa to 45°N in northern Hokkaido. Although there are few K. septemlobus pollen records available, as it produces small amounts of pollen corresponding to an animal-mediated pollination system (Fujimori et al., 2006b, Sakaguchi et al. (2010) reconstructed the LGM distribution of this species using ecological niche modelling. This study provided evidence that the distribution of the species at that time was significantly different from present, with large and continuous range in southern Japan, whereas the species was absent from most of its current northern range except for disjunct northern refugia located on the expanded coastal shelf on the Sea of Japan and Pacific coast sides of Honshu (ca. 38–40°N) (Figure 1b2). These coastal northern refugia may have provided the source for the northward range expansion of the species during the postglacial (Sakaguchi et al., 2010). Given this postglacial migration scenario, the accumulated effects of bottlenecks should have decreased the genetic diversity of newly established populations in northernmost Honshu and Hokkaido (>40°N), where genetic diversity can be expressed as a function of population accessibility to the expansion source.

(a) Locations of the 46 sampled populations of K. septemlobus in the Japanese Archipelago. (b) Predicted distribution probability is shown in each 30-arc-second pixel, based on the ecological niche model for this species at present (b1) and at the last glacial maximum based on the global coupled climate model simulations CCSM3.0 (b2), modified from Sakaguchi et al. (2010).
The aim of this study was to combine information from nuclear microsatellite variation and the species palaeodistribution during the LGM predicted by ecological niche modelling to reveal how glacial survival and postglacial colonisation have shaped the genetic diversity and structure of an important component of temperate deciduous forest, K. septemlobus, in the Japanese Archipelago. In particular, we focus on the respective roles of population bottlenecks and lineage admixture in shaping the genetic structure of new populations during the northward range expansion. First, we describe the geographic distribution of the genetic diversity and the intraspecific lineages in extant populations. The population genetic diversity will then be statistically combined with the reconstructed palaeodistribution to examine the effects of the LGM distribution on the genetic diversity of extant populations. The spatial scales of accessibility to the LGM distribution best relating the two genetic diversity measures (allelic richness and gene diversity) will be discussed in relation to their sensitivity to population bottlenecks and lineage admixture events.
Materials and methods
Plant sampling
We sampled 46 wild populations of K. septemlobus throughout its distribution range in the Japanese Archipelago, that is: in all, 13 populations in Hokkaido (HK1-13), 26 populations in Honshu (HN1-26), 2 in Shikoku (SK1 and 2), 3 in Kyushu (KS1-3) and 1 population each in Yaku (YK) and Okinawa (OK) (Figure 1a). The average sample size per population was 34 (range: 14–79), and 1565 individual trees were sampled in total (Supplementary Table 1). Leaves, or in some cases, winter buds or inner bark, were collected and stored at −20 °C until DNA extraction.
DNA extraction and microsatellite genotyping
Total genomic DNA was extracted from ca. 1.0 cm2 of plant tissues using a modified CTAB method (Murray and Thompson, 1980). Six microsatellite loci (KP20, KP22, KP24, KP26, KP27 and KP28) developed for K. septemlobus (Fujimori et al., 2006a) were used in this study to determine tree genotypes. The microsatellite loci were amplified by PCR under the conditions described by Fujimori et al. (2006a) using a DNA thermal cycler (Dice Model TP600; TaKaRa, Otsu, Japan). The amplified products were loaded onto an auto sequencer (3100 Genetic Analyser; Applied Biosystems, USA), and the fragment lengths were determined using GENOTYPER software (Applied Biosystems, Carlsbad, CA, USA).
Data analysis
Calculation of genetic diversity
To evaluate allelic polymorphism at each microsatellite locus, four genetic diversity statistics below were calculated using FSTAT 2.9.3 software (Goudet, 1995), that is: A, number of alleles per locus; HO, observed heterozygosity within populations; HS, average heterozygosity within populations (Nei, 1987); HT, total genetic diversity across populations. In addition, the fixation index (FIS) was calculated for each locus, and significant departures from Hardy–Weinberg equilibrium were tested by 1000 randomisations using FSTAT 2.9.3.
The level of within-population genetic diversity was evaluated by calculating five genetic diversity statistics using FSTAT 2.9.3, that is: average number of alleles per locus (A), allelic richness (AR, ElMousadik and Petit, 1996), average number of private alleles per locus (PA), observed heterozygosity (HO) and expected heterozygosity or gene diversity (HE, Nei, 1987). When calculating allelic richness using rarefaction method, which can be used to compare allele diversity between populations with different sizes, the standard sample size was set to 14 individuals, or 28 gene copies, which was the smallest population sample size (OK sample). The extent of deviation from Hardy–Weinberg equilibrium was evaluated using FIS estimates (Wright, 1951), and the significance of these values was subsequently tested using the randomisation procedure provided by FSTAT 2.9.3.
Geographic trends in population genetic diversity
We compared levels of genetic variation (AR, HO, HE, FIS and FST) between four island groups defined as geographic sectors, that is Hokkaido (HK1-13), Honshu (HN1-39), Shikoku (SK1, 2) and Kyushu (KS1-3). Regional genetic diversity and two-sided P-values after 1000 permutations to test the effect of population grouping were accessed using FSTAT 2.9.3. In addition, to graphically present spatial trends in population genetic diversity, interpolating surfaces were generated with the inverse distance weighted method incorporated in ArcGIS 9.2 Desktop (ESRI, Redlands, CA, USA) for the diversity statistics of allelic richness and gene diversity. In this interpolation, population OK was excluded, which is isolated from the other main islands and had diversity values much lower than those at other sites. Spatial autocorrelation of population genetic diversity was evaluated as a spline correlogram of the correlation coefficient Moran's I with pointwise confidence intervals (95%), using R-package ncf (Bjornstad, 2009) in R 2.8.1 (R Development Core Team, 2008).
Examining allelic richness components to detect allele capture during lineage admixture
As a clear distinction in allele frequencies was observed between the populations in Sea of Japan and Pacific coasts of northern Honshu from a preliminary data analysis, we hypothesised that lineage admixture during northward postglacial expansion towards north played an important role in shaping the genetic structure in this region. When genetically diverged lineages converge and become gradually admixed during their northward journey, there may be the alleles private to each lineage, which is also present in the populations of admixed origin. To compare allelic constitution in these three populations groups, we decomposed allelic richness (AR, ElMousadik and Petit, 1996) of each group to estimate the components, as denoted here; Common AR (AR common to all the groups), Private AR (AR private to each group), Sea of Japan AR and Pacific AR: AR shared by admixed group with Sea of Japan and Pacific side group, respectively. The three population groups were defined according to the population membership to gene pools estimated by model-based clustering methods and their geographic locations, as (a) Sea of Japan lineage represented by individuals in HN9 and HN10, (b) Pacific lineage by HN13 and (c) individuals of admixed origin by HN3–HN8. We used the rarefaction method (Hurlbert, 1971) to calculate the expected number of alleles found when 98 genes copies, equal to the minimum number of gene copies in Sea of Japan lineage group, are sampled at the locus in question. We then summed the expected number of alleles for each allelic richness component, and finally averaged these components across the six loci.
Genetic differentiation and isolation by distance analysis
The common genetic differentiation indices FST (Weir and Cockerham, 1984) was also calculated in this study. Significant genetic differentiation in FST was tested by 1000 randomisations using FSTAT 2.9.3. However, when using highly polymorphic genetic markers, the level of homozygosity must decrease, leading to significant underestimations of genetic differentiation (Nei, 1987; Hedrick, 1999). To allow comparisons of differentiation levels among different genetic markers, the standardised genetic differentiation index G′ST (Hedrick, 2005) and the actual differentiation D (Jost, 2008) were calculated using MSA 4.05 software (Dieringer and Schlotterer, 2003) and SMOGD 1.2.5 (Crawford, 2010), respectively. All these differentiation indices were calculated using all the population data and the data excluding the OK population.
Relationships between pairwise genetic distances and log-transformed geographic distances between populations were analysed via Mantel tests with 999 permutations provided in GenAlEx 6.1 (Peakall and Smouse, 2006). Pairwise FST/(1–FST) values between populations were employed for genetic distance, as suggested by Rousset (1997). We first analysed (a) all 46 populations and subsequently analysed (b) 45 populations excluding the OK population, as the pairwise genetic distances of this population were apparently larger than those of the others, (c) populations in Honshu, and (d) in Hokkaido.
Genetic structure in the Japanese Archipelago
Distance-based population clustering
Pairwise genetic distances of Nei's DA (Nei et al., 1983), with which a higher probability of obtaining a correct phylogenetic tree topology can be attained (Takezaki and Nei, 1996), were calculated among the 46 populations to construct a neighbour-joining tree (Saitou and Nei, 1987) using Populations 1.2.30 beta software (Langella, 1999). The application of a simple branching tree model to data, however, can be problematic when systematic errors exist, such as inappropriate assumptions in the evolutionary model and sampling errors resulting from small numbers of observed loci (Huson and Bryant, 2006). Thus, to explore and graphically present these ambiguities, we also constructed a split network (Bryant and Moulton, 2004) on the basis of a distance matrix of Nei's DA (Nei et al., 1983) using SplitsTree4 ver. 4.10 (Huson, 1998; Huson and Bryant, 2006). In addition, a principal component analysis using a pairwise FST matrix was conducted with the aid of GenAlEx 6.1 (Peakall and Smouse, 2006).
Model-based clustering
We employed two model-based Bayesian algorithms implemented in STRUCTURE 2.3 and TESS 2.3.1 (Pritchard et al., 2000; Durand et al., 2009) to detect genetic structure within the collected samples. For STRUCTURE analysis, the assumption was made that each individual had admixed ancestral origins that different gene pools retained their allele frequency correlated (‘correlated allele frequencies model’ in Falush et al. (2003)), and that the sampling locations are designated as previous information to obtain better parameter estimates (Hubisz et al., 2009). Ten individual simulations were run for each K (K=1–15), with 20 000 burn-in steps followed by 10 000 Markov chain Monte Carlo steps. Pritchard et al. (2009), however, pointed out that data sets in real world do not conform precisely to the non-spatial STRUCTURE model (for exampe, due to isolation by distance). In such cases, the inferred number of gene pools (K), and the corresponding allele frequencies in each pool can be rather arbitrary. Recently, Durand et al. (2009) proposed a spatial hierarchical Bayesian algorithm implemented in TESS 2.3.1, in which individual membership can be modelled as the sum of spatial components. We estimated the population structure within the samples using this algorithm by incorporating individual geographic covariates in the prior distributions on the admixture memberships. Ten individual simulations were run for each Kmax (Kmax=2–15), with 10 000 burn-in steps followed by 20 000 Markov chain Monte Carlo steps.
Relationship between the LGM distribution and genetic diversity of extant population
We analysed two different genetic diversity measures (allelic richness and arcsine-transformed gene diversity) of the current populations as a function of accessibility to the LGM palaeodistribution to explore whether there is non-equilibrium state in spatial pattern of genetic diversity due to postglacial range expansion, a similar situation in tree species diversity of Europe (Svenning, 2007). Population accessibility index was expressed as summed distribution probability of K. septemlobus during the LGM, reconstructed by Sakaguchi et al. (2010). The reconstructed distribution probability given for each 30-sec pixel within the study area was sampled and summed within multiple-ring buffers generated from the sampled locations using ArcGIS 9.2 Desktop (ESRI). Hereafter, this summed distribution probability will be called ‘accessibility’ to the palaeodistribution. The sizes of sampling radius were varied from 100 km to 2000 km in 100-km increments as estimated effect of the fixed term may vary according to the spatial scale with which accessibility is evaluated. We used a linear mixed model with the loci as random intercepts, because diversity values at these loci are likely to be more related to each other than to the values from different loci. Our hypotheses were then translated into 21 candidate models, including full models with the fixed term ‘accessibility’ evaluated using different 20 spatial scales and a null model. To explore the spatial scale best explaining variations in each genetic diversity index, we selected the most parsimonious model with minimum Akaike's information criterion value among the candidates. We used R-package nlme by Pinheiro et al. (2010) in R 2.8.1 (R Development Core Team, 2008) to obtain parameter estimates by the restricted maximum likelihood method.
Results
Genetic polymorphisms overall and within populations
The number of alleles observed per locus ranged from 18 to 29, with an average of 22.7 alleles; a total of 136 alleles were scored (Table 1). The total genetic diversity over all populations (HT), observed diversity within populations (HO) and expected diversity within populations (HS) were 0.853, 0.679 and 0.781, respectively. Significant departures from Hardy–Weinberg equilibrium were detected at all loci except for KP24. When genetic diversity indices were compared between island groups, significant differences were detected in allelic richness and expected heterozygosity with the highest level in Shikoku and lowest in Hokkaido (Table 2). Fourteen populations on Honshu, Shikoku, Kyushu and Okinawa Islands harboured one to three private alleles, whereas all 13 populations from Hokkaido contained no private alleles (Supplementary Table 1). The population in far-isolated Okinawa Island had the lowest gene diversity and a highly significant FIS value.
Significant positive spatial autocorrelations over a geographic distance of 250 km were observed for allelic richness and gene diversity (Figures 2a and b). In addition, negative spatial autocorrelations were detected for allelic richness between distance classes from 1200–1500 km, and for gene diversity from 1200–1600 km. The interpolation of allelic richness value (Figure 2a) illustrated that the bulk of genetic diversity was centred on westernmost Honshu, Shikoku and the northern part of Kyushu. From these areas of high diversity, allelic richness decreased significantly towards the northern part of Honshu and southern part of Kyushu. Within the island of Hokkaido, allelic richness decreased rapidly from the southwest to the northeast. A similar geographic trend was evident for gene diversity (Figure 2b), with a centre of diversity in the southwest of the Japanese Archipelago and decreasing trend towards northeastern Hokkaido. However, in contrast to allelic richness, a second centre of gene diversity was observed in the northernmost part of Honshu. The existence of these two centres of gene diversity, located about 1000 km from each other, was supported by a marginally significant negative spatial autocorrelation for gene diversity at the distance of 500 km in the correlogram.

Spatial organisation of the genetic diversity of K. septemlobus populations in the Japanese Archipelago (a: allelic richness, b: gene diversity). Diversity estimates for 45 populations excluding the Okinawa population were used to generate the interpolating surface using the inverse distance weighted method. Spatial autocorrelation is addressed as spline correlogram of the correlation coefficient, Moran's I, with pointwise confidence intervals (95%) superimposed.
Allelic components in population groups of the admixture zone
Genetic diversity (AR and HE) was observed to be highest in the admixed group among the three population groups in the northernmost Honshu (Table 3), although it is located farthest from the reconstructed glacial ranges of K. septemlobus (Figure 1b2). The admixed group was also shown to share 14.2 and 12.0% of allelic richness only with Sea of Japan and Pacific coast groups, respectively. This allelic richness component (26.2% in total) was much larger than private allelic richness (9.3%) in the admixture zone.
Genetic differentiation between populations
Genetic differentiation between populations, as measured by global FST, was significant at all six loci, ranging from 0.045 to 0.078, with an average of 0.063 (Table 1). The average G’ST and D was estimated at 0.402 and 0.358, respectively, which were much higher than the GST (average: 0.085), indicating that GST was underestimated due to the high polymorphism of the microsatellite markers used in this study. When genetic differentiation was evaluated without the OK population, differentiation level became slightly lower. Pairwise genetic distances measured as FST/(1–FST) were significantly correlated with the pairwise geographic distances between all populations (P<0.01, r=1.668e–2, R2=0.156; Figure 3a). Genetic distances between the OK population and other populations were higher (>0.15) than those between other pairs, so that the slope of the regression line became gentler when the outlier was excluded from the analysis (P<0.01, r=0.606e–2, R2=0.124; Figure 3b). Isolation by distance analysis conducted between Honshu populations detected significant correlation (P<0.01, r=0.351e–2, R2=0.051; Figure 3c), whereas no significant correlation was detected between genetic and geographic distances between Hokkaido population pairs (P=0.14, R2=0.021; Figure 3d).

The relationship between the pairwise genetic and geographic distance matrices. The effect of isolation by distance was estimated for all population pairs (a), for pairs without the Okinawa population pairs (b), for pairs in Honshu Island (c), and in Hokkaido Island (d).
Spatial genetic structure in the Japanese Archipelago
The neighbour-joining tree on the basis of genetic distance of Nei's DA (Nei et al., 1983) showed gradual genetic differentiation along the archipelago, that is, a north–south differentiation pattern (Supplementary Figure 1a). Populations in Hokkaido were more closely related to the northern Sea of Japan populations (HN1, 2, 4, 9, 10) than to the northern Pacific Ocean populations (HN5, 6, 7, 13) of Honshu, although the two population groups are almost equally distant from Hokkaido. The length of the terminal clade of the population on Okinawa was estimated to be much longer than those of other populations, suggesting considerable genetic differentiation of this insular population from the main islands. The population network included many boxes and appeared to be distinctly non-tree-like (Supplementary Figure 1b), meaning ambiguity in applying a simple tree model to the data, although the relationship between populations was almost the same as those shown in the neighbour-joining tree. Principal component analysis conducted for the populations in main islands appeared to reflect a north–south differentiation pattern on the first axis, as suggested from neighbour-joining tree and population network, and a west–east differentiation between Sea of Japan and Pacific coast populations in northern Honshu on the second axis (Supplementary Figure 2).
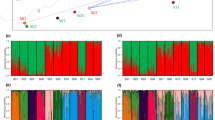
In the model-based clustering STRUCTURE analysis, the log likelihood of the data continued to increase logarithmically as the number of gene pools (K) increased from 1 to 15 (data not shown). As the spatial distribution of the gene pools from K=2 to K=6 apparently showed positive spatial autocorrelation patterns, we considered these cluster assignments to be biologically meaningful. When two gene pools were assumed, we found a gradual admixing along the archipelago (Figure 4b). The northern gene pool, coloured in blue, dominated in the individuals sampled in Hokkaido, whereas the southern cluster (red) appeared mainly in western Japan. In the northern part of Honshu, the two gene pools extensively admixed. The average genetic distance (expected heterozygosity) between individuals in the same cluster was slightly larger in the southern (0.87) compared with the northern (0.80) pool, and the northern pool was estimated to have experienced stronger genetic drift away from the ancestral pool (FST=0.081) than the southern pool (FST=0.015). In the clustering with K=3, another (yellow) gene pool was assigned to the individuals in the northern part of Honshu, where high admixture of the two gene pools were inferred in the K=2 clustering. This gene pool appeared to be more dominant in Pacific Ocean populations (HN5–7, HN13) than in the Sea of Japan populations (HN1, 2, 4, 9, 10). When we set the number of gene pools to four, some of the blue pool detected in K=3 were reassigned to a new gene pool (light blue), which dominated along the Sea of Japan-side populations in Honshu. From K=5–6, new gene pools were shown to dominate in the following areas: K=5, green pool in the central HN populations (that is, HN17–HN23); K=6, light green pool in the YK and OK populations. Although the extent of genetic admixture between populations appeared to be slightly larger than estimated by STRUCTURE analysis, a spatial hierarchical Bayesian clustering TESS analysis produced a similar genetic clustering pattern of populations with non-spatial STRUCTURE analysis (Supplementary Figure 3), suggesting that any spurious clustering in STRUCTURE analysis caused by spatial autocorrelation structure in the data is unlikely.

(a) Coefficients of individual membership to gene pools estimated by the model-based clustering method of Pritchard et al. (2000). Clustering patterns are shown for number of gene pools (K=2–6). (b) Geographic variation in population-level membership to gene pools (K=2–4) are superimposed on the study area map. For each cluster, the expected heterozygosity within each pool is shown by the relative sizes of coloured circles, FST values representing the genetic drift undergone by each pool independent of the ancestral allele frequencies and the neighbour-joining tree related to the gene pools are presented.
Effects of accessibility to the LGM distribution on extant population genetic diversity
Allelic richness and gene diversity were modelled as a function of population accessibility to the LGM palaeodistribution. For allelic richness, the Akaike's information criterion value of the model with a sampling scale of 1000 km was the lowest among the candidate models. In the allelic richness model (d.f.=269), the fixed term ‘accessibility’ had significant positive effects on allelic richness (t=8.213, P<0.001; Figure 5a). The model reconstructed the highest level of allelic richness among the populations in western Honshu and Shikoku, where large areas of suitable habitat existed during the LGM. As the populations become further remote from these areas, allelic richness was predicted to decrease, suggesting a ‘centre-peripheral pattern’ dependant on the LGM distribution (interpolated surface of accessibility index is available in Supplementary Figure 4a). The spatial scale of the gene diversity model (d.f.=269) best explaining genetic variation was shown to be 1700 km. Accessibility to the LGM range was also shown to significantly increase gene diversity (t=6.780, P<0.001). Contrary to the predicted allelic richness, the gene diversity model reconstructed a simple spatial organisation of genetic diversity, that is, a ‘northern purity and southern richness’ pattern.

Genetic diversity values are plotted against population accessibility to the LGM palaeodistribution in the Japanese Archipelago (a: allelic richness, b: gene diversity). Observation values at six microsatellite loci are indicated by different symbols. The predictor, standardised accessibility, was evaluated by ring buffers with diameters of 1000 km for allelic richness and 1700 km for gene diversity. Fitted values (solid line) and 95% confidence intervals (dashed line) for the linear mixed model are also superimposed.
Discussion
Contrasting histories of southern and northern populations on the main islands
Populations in southwestern Japan, most notably in westernmost Honshu and Shikoku, were characterised by the highest levels of genetic diversity (allelic richness and gene diversity) and by the greatest number of private alleles. In addition, STRUCTURE analysis detected the most ancestral and genetically diverse gene pool in these areas (Figure 4). These lines of genetic evidences suggest that populations have persisted in these areas for a long time, including through the LGM, and is consistent with the predicted palaeodistribution of the species that showed high probability of occurrence in lowland areas of southern Japan during the LGM (Sakaguchi et al., 2010). Thus, K. septemlobus is likely to have survived in the locations that largely overlaps with the suggested refugial areas for other temperate forest trees, such as Fagus crenata and Cryptomeria japonica based on palynological and phylogeographic evidences (Tsukada, 1982a, 1982b, 1986; Yasuda, 1985). Considering K. septemlobus is currently confined to higher altitudes (>300 m a.s.l.) in southwestern Japan, the species probably maintained its presence by elevating its range into nearby mountains at the onset of climate warming during the postglacial period. The high genetic diversity, evidence for lack of genetic drift as indicated by low Fst values in STRUCTURE analysis, and private alleles in these populations indicate that genetic bottlenecks were not severe during any local altitudinal migrations (Hewitt, 1996).
According to the prediction from the ecological niche model, in contrast to southern areas, Hokkaido should not have provided suitable environments for K. septemlobus during the LGM (Figure 1b2). Abundant fossil pollens and spores of cold-tolerant coniferous species (Pinus, Picea and Larix) and Betula spp. accompanied by herbaceous species of Cyperaceae and Lycopodiaceae during this period indicate that open taiga vegetation associated with mires developed across most of Hokkaido during that period (Morita, 1985; Igarashi, 1993). The climatic conditions under which such taiga vegetation develops are likely too severe for a temperate species like K. septemlobus to survive, and it is currently absent from such vegetation. In line with this historical scenario, our genetic analysis detected no evidence that the species persisted in Hokkaido during the last glacial period, instead of being most consistent with a recent expansion into this territory some time after the LGM. Firstly, the genetic diversity of the populations in Hokkaido was the lowest of the four main islands and no private alleles were detected. The predominant lineage detected by STRUCTURE analysis had consistently larger FST values than observed in the other lineages, indicating that it had experienced sequential genetic drift from the ancestral populations. Secondly, despite the current widespread range of the species in Hokkaido, no significant relationship between genetic and geographic distances was observed. This could be interpreted as evidence for a non-equilibrium situation where the species has recently colonised its current distribution (Slatkin, 1993). The clear decrease in genetic diversity from southwestern to northeastern parts of the island is concordant with postglacial colonisation that started in the southwest, which probably synchronised with the directional postglacial migration inferred for other deciduous broadleaf forest species in Hokkaido (ca. 8000 years BP, reviewed in Yamada, 1998). During this colonisation, the founding lineage would have divided into two groups to proceed around the central high mountains towards the northeastern part of this island and would have gradually lost genetic diversity with each long-distance colonisation (Le Corre and Kremer, 1998). Given that Hokkaido was colonised by lineages passing through Tsugaru Strait from Honshu during the Holocene, the number of immigrants from the Sea of Japan side may have overwhelmed those from the Pacific side because the gene pool predominating along the Sea of Japan side of northern Honshu was detected in individuals at the far greatest frequency in Hokkaido. Chloroplast and nuclear genetic lineages that characterise Sea of Japan-side populations of F. crenata are also predominant in Hokkaido populations (Fujii et al., 2002; Hiraoka and Tomaru, 2009). This concordance among two temperate tree species may indicate that the northward range expansion of the temperate forest biome along the Sea of Japan side preceded the expansion along the Pacific side in Honshu.
High gene diversity in northernmost Honshu
Exceptionally high levels of gene diversity within the species were detected in the populations in northernmost Honshu, where vast areas of unsuitable habitat for K. septemlobus were predicted during the LGM (Sakaguchi et al., 2010). This apparent contradiction between genetic and modelling evidence was further confirmed by the accessibility analysis, where population accessibility to the LGM palaeodistribution failed to predict the diversity centre in the northernmost Honshu, suggesting that factors other than simple range expansion from core refugia may have influenced genetic diversity in this area. Distinct intraspecific lineages were found to be distributed along the Pacific and Sea of Japan sides and extensively admixed in northernmost Honshu. Similar lineage distribution patterns are also reported in F. crenata and C. japonica in the same area (Fujii et al., 2002; Tsumura et al., 2007; Hiraoka and Tomaru, 2009). Genetic divergence between these two lineages would likely reflect the separation of coastal refugia by the central mountain ranges during past glacial periods, including the LGM. As the climate became warmer after the LGM, these lineages would likely have begun to expand from both coasts and eventually admixed in the northernmost part of the island. Admixture of genetically differentiated populations would have increased the genetic heterogeneity of the newly established populations, comparable to the populations in southern Japan. Similar observations of elevated genetic diversity in admixed zones have been reported in other wind-pollinated tree species, for example, F. sylvatica, Fraxinus excelsior, Pinus resinosa (Comps et al., 2001; Walter and Epperson, 2001; Petit et al., 2003a; Heuertz et al., 2004).
Although lineage admixture would be the most influential process that elevated genetic diversity in the northernmost Honshu, cryptic refugia during the LGM may have played important roles in maintaining the genetic diversity there. In this study, we found that some populations in the northernmost Honshu, including both the Sea of Japan and Pacific-side populations, possess private alleles. Apart from the potential for post LGM evolution of these alleles, this genetic data provides new evidence that K. septemlobus was able to persist further north, at least to 40oN, on the Pacific coast of Japan than was predicted by ecological niche modelling. Recent phylogeographic studies on temperate trees and animals in Japan suggested that small populations may have survived in the northernmost part of Honshu, separated from the southern large glacial refugia (Tsuda and Ide, 2005; Tsumura et al., 2007; Ohnishi et al., 2009).
Spatial scales relating palaeodistribution during the ice age and extant population genetic diversity
We showed positive correlation between population genetic diversity and its accessibility to surrounding LGM distribution probability, at larger spatial scale for gene diversity than allelic richness. It is probable that the rates of diversity loss due to bottlenecks in long-distance dispersals during the postglacial are likely to have influenced the selection of the spatial scales in this analysis. As genetic drift eliminates low-frequency alleles easily, the number of alleles decreases faster than gene diversity (Nei et al., 1975; Maruyama and Fuerst, 1984). Accordingly, the smaller spatial scale selected for allelic richness model can be explained by a loss in allelic diversity caused by sequential of bottlenecks occurring over shorter distance from the leading edges of the LGM populations, compared with gene diversity. Another non-exclusive explanation may involve the lineage admixture event that occurred in the northernmost Honshu. When lineage admixture increases gene diversity more strongly than allelic richness in established tree populations (Comps et al., 2001; Widmer and Lexer, 2001), elevated gene diversity due to lineage admixture might lead to an underestimation of the effects of bottlenecks during range expansion as compared with a simple situation without admixture. This masking effect of lineage admixture on the rate of genetic diversity loss may have increased the spatial scale selected for the gene diversity model.
Strong isolation effect of straits on small insular populations
The level of genetic differentiation between the OK and other populations was much higher than that expected from the isolation by distance model deduced from mainland populations (Figures 3a and b). Environmental heterogeneity (that is, the wide strait between the OK and the YK populations) likely reduced any animal-mediated seed and pollen dispersal, accelerating genetic differentiation in this isolated population. As STRUCTURE analysis detected a common gene pool (light-green above K=6) between the OK and YK populations, individuals are likely to have migrated from Yaku to Okinawa Island and established the populations there. The low level of genetic diversity in the OK population may reflect strong founder effect at those times. During the Quaternary period, the island chain from Yaku to Taiwan repeatedly formed landbridges due to sea-level changes induced by climatic oscillations (Kimura, 1996, 2000). Cardiandra plants (Hydrangeaceae) on these islands were shown to have a common ancestor, suggesting that their range expanded along the landbridge, passing from Taiwan to Japan during glacial periods (Setoguchi et al., 2006). To clarify the origin and population history of the OK population of K. septemlobus, further phylogeographic study including samples from mainland China and the Korean Peninsula should be conducted in the future.
Conclusions
The combined evidence from nuclear microsatellites and ecological niche modelling clearly illustrated that climate-driven range fluctuations have shaped population genetic diversity and structure of K. septemlobus in the Japanese Archipelago. Genetic diversity cline from the south to north was well predicted by population accessibility to the LGM palaeodistribution probability, suggesting spatial arrangement of glacial refugia controlled overall picture of genetic diversity. However, at the regional scale, we found that local population dynamics played more important roles in determining the level of population genetic diversity. Another centre of genetic diversity detected in the northernmost Honshu in addition to southern Japan is likely to have been generated by genetic admixture during the postglacial range expansion of two distinct lineages, probably having sheltered in cryptic northern refugia on the exposed coastal landscapes of Pacific and Sea of Japan sides. During long-distance colonisation towards north, accumulated bottleneck effects are likely to have decreased allelic richness at a greater rate than gene diversity. The fact that variation in allelic richness among populations was best explained at relatively smaller spatial scale than gene diversity may reflect such differences in sensitivity of genetic diversity indices in the face of population bottlenecks.
References
Austerlitz F, Garnier-Gere PH (2003). Modelling the impact of colonisation on genetic diversity and differentiation of forest trees: interaction of life cycle, pollen flow and seed long-distance dispersal. Heredity 90: 282–290.
Bjornstad ON (2009). ncf: Spatial nonparametric covariance functions. R package version 1.1-3.
Bryant D, Moulton V (2004). Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21: 255–265.
Carstens BC, Richards CL (2007). Integrating coalescent and ecological niche modeling in comparative phylogeography. Evolution 61: 1439–1454.
Comps B, Gomory D, Letouzey J, Thiebaut B, Petit RJ (2001). Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157: 389–397.
Crawford NG (2010). SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10: 556–557.
Dieringer D, Schlotterer C (2003). MICROSATELLITE ANALYSER (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3: 167–169.
Durand E, Jay F, Gaggiotti OE, Francois O (2009). Spatial inference of admixture proportions and secondary contact zones. Mol Biol Evol 26: 1963–1973.
ElMousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L) Skeels endemic to Morocco. Theor Appl Genet 92: 832–839.
Falush D, Stephens M, Pritchard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
Fujii N, Tomaru N, Okuyama K, Koike T, Mikami T, Ueda K (2002). Chloroplast DNA phylogeography of Fagus crenata (Fagaceae) in Japan. Plant Syst Evol 232: 21–33.
Fujimori N, Kenta T, Kond T, Kameyama Y, Isagi Y, Nakashizuka T (2006a). Microsatellite loci for a sparsely distributed species, Kalopanax pictus (Araliaceae) in Japanese temperate forests. Mol Ecol Notes 6: 468–469.
Fujimori N, Samejima H, Kenta T, Ichie T, Shibata M, Iida S et al. (2006b). Reproductive success and distance to conspecific adults in the sparsely distributed tree Kalopanax pictus. J Plant Res 119: 195–203.
Gao LM, Moeller M, Zhang XM, Hollingsworth ML, Liu J, Mill RR et al. (2007). High variation and strong phylogeographic pattern among cpDNA haplotypes in Taxus wallichiana (Taxaceae) in China and North Vietnam. Mol Ecol 16: 4684–4698.
Goudet J (1995). FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Hedrick PW (1999). Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53: 313–318.
Hedrick PW (2005). A standardized genetic differentiation measure. Evolution 59: 1633–1638.
Heuertz M, Hausman JF, Hardy OJ, Vendramin GG, Frascaria-Lacoste N, Vekemans X (2004). Nuclear microsatellites reveal contrasting patterns of genetic structure between western and southeastern European populations of the common ash (Fraxinus excelsior L). Evolution 58: 976–988.
Hewitt G (2000). The genetic legacy of the Quaternary ice ages. Nature 405: 907–913.
Hewitt GM (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linnean Soc 58: 247–276.
Hiraoka K, Tomaru N (2009). Genetic divergence in nuclear genomes between populations of Fagus crenata along the Japan Sea and Pacific sides of Japan. J Plant Res 122: 269–282.
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9: 1322–1332.
Hurlbert SH (1971). Nonconcept of species diversity—critique and alternative parameters. Ecology 52: 577.
Huson DH (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14: 68–73.
Huson DH, Bryant D (2006). Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267.
Igarashi Y (1993). Vegetation history of Kenbuchi basin and Furano basin in Hokkaido, north Japan, since 32 000 yrs BP. Quat Res (Jap) 32: 89–105.
Jost L (2008). GST and its relatives do not measure differentiation. Mol Ecol 17: 4015–4026.
Kimura M (1996). Quaternary paleogeography of the Ryukyu Arc. J Geogr 105: 259–285.
Kimura M (2000). Paleogeography of the Ryukyu Islands. Tropics 10: 5–24.
Langella O (1999) Populations, 1.2.30 CNRS UPR9034.
Le Corre V, Kremer A (1998). Cumulative effects of founding events during colonisation on genetic diversity and differentiation in an island and stepping-stone model. J Evol Biol 11: 495–512.
Lisiecki LE, Raymo ME (2005). A Plio-Pleistocene stack of 57 globally distributed benthic δ18 O records. Paleoceanography 20 (PA1003).
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gomory D et al. (2006). A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171: 199–221.
Maruyama T, Fuerst PA (1984). Population bottlenecks and nonequilibrium models in population-Genetics. 1. allele numbers when populations evolve from zero variability. Genetics 108: 745–763.
McLachlan JS, Clark JS, Manos PS (2005). Molecular indicators of tree migration capacity under rapid climate change. Ecology 86: 2088–2098.
Morita Y (1985). Palynological study of the deposits from the Uryu Numa Moor in Mt. Shokanbetsu, Hokkaido. Ann Tohoku Geogr Assoc 37: 166–172.
Murray MG, Thompson WF (1980). Rapid isolation of high molecular-weight plant DNA. Nucleic Acids Res 8: 4321–4325.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Nei M, Maruyama T, Chakraborty R (1975). Bottleneck effect and genetic-variability in populations. Evolution 29: 1–10.
Nei M, Tajima F, Tateno Y (1983). Accuracy of estimated phylogenetic trees from molecular-data .2. gene-frequency data. J Mol Evol 19: 153–170.
Ohashi H (1994). Nomenclature of Kalopanax septemlobus (Thunb. ex Murray) Koidzumi and classification of its infraspecific taxa (Araliaceae). J Jap Bot 69: 28–31.
Ohba H (1999). Family Araliacecae. In: Iwatsuki, K, Boufford, DE and Ohba, H (eds). Flora of Japan. Kodan-sha Tokyo. Vol. IIc, pp 259-267.
Ohnishi N, Uno R, Ishibashi Y, Tamate HB, Oi T (2009). The influence of climatic oscillations during the Quaternary Era on the genetic structure of Asian black bears in Japan. Heredity 102: 579–589.
Peakall R, Smouse PE (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Peterson AT, Martinez-Meyer E, Gonzalez-Salazar C (2004). Reconstructing the Pleistocene geography of the Aphelocoma jays (Corvidae). Divers Distrib 10: 237–246.
Petit JR, Jouzel J, Raynaud D, Barkov NI, Barnola JM, Basile I et al. (1999). Climate and atmospheric history of the past 4 20 000 years from the Vostok ice core, Antarctica. Nature 399: 429–436.
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al. (2003a). Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565.
Petit RJ, Csaikl UM, Bordacs S, Burg K, Coart E, Cottrell J et al. (2003b). Chloroplast DNA variation in European white oaks phylogeography and patterns of diversity based on data from over 2600 populations (vol 156, pg 5, 2002). For Ecol Manage 176: 595–599.
Pinheiro J, Bates D, DebRoy S, Sarkar D, Team tRDC (2010). nlme: Linear and nonlinear mixed effects models. R package version 3: 1–97.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Pritchard JK, Wen X, Falush D (2009). Documentation for structure software: Version 2.3. http://pritchbsduchicagoedu/structure_software/release_versions/v232/structure_docpdf.
R Development Core Team (2008) R version 2.8.1.
Rousset F (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Saitou N, Nei M (1987). The neighbor-joining method—a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425.
Sakaguchi S, Sakurai S, Yamasaki M, Isagi Y (2010). How did the exposed seafloor function in postglacial northward range expansion of Kalopanax septemlobus? Evidence from ecological niche modelling. Ecol Res 25: 1183–1195.
Setoguchi H, Yukawa T, Tokuoka T, Momohara A, Sogo A, Takaso T et al. (2006). Phylogeography of the genus Cardiandra based on genetic variation in cpDNA sequences. J Plant Res 119: 401–405.
Slatkin M (1993). Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47: 264–279.
Solomon SE, Bacci M, Martins J, Vinha GG, Mueller UG (2008). Paleodistributions and comparative molecular phylogeography of leafcutter ants (Atta spp.) provide new insight into the origins of Amazonian diversity. PLoS One 3: e2738.
Svenning J-C, Normand S, Kageyama M (2008). Glacial refugia of temperate trees in Europe: insights from species distribution modelling. J Ecol 96: 1117–1127.
Svenning JC (2007). Could the tree diversity pattern in Europe be generated by postglacial dispersal limitation? Ecol Lett 10: 453–460.
Takezaki N, Nei M (1996). Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144: 389–399.
Tomaru N, Mitsutsuji T, Takahashi M, Tsumura Y, Uchida K, Ohba K (1997). Genetic diversity in Fagus crenata (Japanese beech): influence of the distributional shift during the late-Quaternary. Heredity 78: 241–251.
Tsuda Y, Ide Y (2005). Wide-range analysis of genetic structure of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in the cool temperate zone of Japan. Mol Ecol 14: 3929–3941.
Tsukada M (1982a). Cryptomeria Japonica—Glacial Refugia and late-Glacial and post-Glacial migration. Ecology 63: 1091–1105.
Tsukada M (1982b). Late Quaternary shift of Fagus distribution. Bot Mag Tokyo 95: 203–217.
Tsukada M (1983). Vegetation and climate during the last glacial maximum in Japan. Quat Res 19: 212–235.
Tsukada M (1985). Map of vegetation during the last glacial maximum in Japan. Quat Res 23: 369–381.
Tsukada M (1986). Altitudinal and latitudinal migration of Cryptomeria japonica for the past 20 000 years in Japan. Quat Res 26: 135–152.
Tsumura Y, Kado T, Takahashi T, Tani N, Ujino-Ihara T, Iwata H (2007). Genome scan to detect genetic structure and adaptive genes of natural populations of Cryptomeria japonica. Genetics 176: 2393–2403.
Waltari E, Hijmans RJ, Peterson AT, Nyári ÁS, Perkins SL (2007). Locating pleistocene refugia: comparing phylogeographic and ecological niche model predictions. PLoS One 2: e563.
Walter R, Epperson BK (2001). Geographic pattern of genetic variation in Pinus resinosa: area of greatest diversity is not the origin of postglacial populations. Mol Ecol 10: 103–111.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population-structure. Evolution 38: 1358–1370.
Widmer A, Lexer C (2001). Glacial refugia: sanctuaries for allelic richness, but not for gene diversity. Trends Ecol Evol 16: 267–269.
Wright S (1951). The genetical structure of populations. Ann Eug 15: 323–354.
Xiang Q, Lowry PP (2007). Genus Kalopanax Flora of China vol. 13. Science Press: Beijing; Missouri Botanical Garden Press: St Louis. p 441.
Yamada G (1998). Vegetation history of Hokkaido I, Northern Hokkaido. In: Yasuda Y, Miyoshi N (eds). The Illustrated Vegetation History of the Japanese Archipelago. Asakura-shoten: Tokyo. pp 39–50.
Yasuda Y (1985). Natural history and civilization of two beech forests in the east and the west. In: Umehara T (ed.). Culture of Beech Forest Zones. Shisaku-sha: Tokyo. pp 29–63.
Acknowledgements
We thank K Niiyama, M Shibata, K Hoshizaki, S Goto, H Samejima, Seiji Higa, S Masutani, S Mori, Y Matsuki, R Goda, C Sumiyoshi, Shinako Higa, Y Matsuura, Y Azuma, Y Isshiki and T Toyama for their great help in collecting plant materials. We are indebted to Y Tsuda and J Worth for their helpful comments on data analysis and manuscript improvement. We are also grateful to the managing editor, Professor R Butlin, and three anonymous reviewers for insightful comments and suggestions in an early version of this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Rights and permissions
About this article
Cite this article
Sakaguchi, S., Takeuchi, Y., Yamasaki, M. et al. Lineage admixture during postglacial range expansion is responsible for the increased gene diversity of Kalopanax septemlobus in a recently colonised territory. Heredity 107, 338–348 (2011). https://doi.org/10.1038/hdy.2011.20
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/hdy.2011.20
Keywords
This article is cited by
-
Genetic differentiation and evolution of broad-leaved evergreen shrub and tree varieties of Daphniphyllum macropodum (Daphniphyllaceae)
Heredity (2023)
-
Inferring historical survivals of climate relicts: the effects of climate changes, geography, and population-specific factors on herbaceous hydrangeas
Heredity (2021)
-
How co-distribution of two related azaleas (Rhododendron) developed in the Japanese archipelago: insights from evolutionary and demographic analyses
Tree Genetics & Genomes (2021)
-
Low seed fertility of hermaphrodites is maintained in a gynodioecious species throughout the distribution range in Japan
Plant Systematics and Evolution (2021)
-
Habitat preference differentiates the Holocene range dynamics but not barrier effects on two sympatric, congeneric trees (Tristaniopsis, Myrtaceae)
Heredity (2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}